Previous--Is Sustainability a Viable Concept in the Management of Confined Aquifers in Kansas? || Next--Yield Estimates for Surface-water Sources
This article available as an Acrobat PDF file (1.2 Mb).
The chemistry of water determines its aesthetic character and imposes limitations for some uses. Damage to the health of animals (e.g., humans or livestock) or plants, damage to infrastructure (e.g., pipes, boilers), damage to soils, or impairment of manufacturing processes are among the potential consequences of using water of poor quality. Availability of ground water, that is, water quantity, is discussed in other chapters in this volume. In this chapter, the processes affecting the chemical quality of ground water are discussed because the quality of water is a critical part of the sustainable yield assessment of an aquifer. Although quantity of water is important and limiting in many cases, unacceptable water chemistry can render even very large available volumes of water useless.
A review of the most important chemical reactions and processes affecting water chemistry in the first section of this chapter provides a foundation for understanding variations in water chemistry, both natural and induced. Ground water is nearly always moving through an aquifer, although rates vary greatly. This movement of ground water flushes out resident fluids. The fluids being flushed can be residual from original deposition of the aquifer, such as sea water, or can be some other type of saline or freshwater from a time when ground-water flow was different from the present. During the flushing process, chemical reactions occur as the new water is chemically altered by the rock: dissolution or precipitation of minerals, attachment (sorption) or detachment (desorption) of ions onto surfaces, or ion exchange may occur. These reactions apply principally to dissolved species that do not change their electrochemical state (redox state) or are not volatile. Those groups of species that do make such changes, and so, in a sense, change form, include nitrogen compounds such as nitrate, organic compounds, and some metals. Part of the section on chemical reactions reviews the conditions that control the reactions involved in this change of form. Furthermore, some dissolved species may act as convenient indicators of different water sources, as they do not tend to participate in chemical reactions. All of this information (the tendency to participate in reactions and knowledge about what types of chemical reactions are likely to occur) becomes helpful in understanding the origins of the chemistry of a ground water (hindsight) and in predicting the consequences of aquifer development on water chemistry (prediction).
The second section of this chapter reviews the pathways by which water with unacceptable quality can enter aquifers. A review of the importance of the unsaturated zone to altering the chemistry of ground-water recharge is followed by discussion of different sources of recharge and how they can be affected by the pumping of ground water. Both intra-aquifer flow (mixing of fluids from down- and updip, for example) and inter-aquifer flow (in which pumping induces cross formational flow of fluids) have the potential to mix waters of different chemistry that are not in equilibrium with the conditions at the pumping welles). Pertinent examples from Kansas and elsewhere illustrate the potential impact of mismanagement of ground-water resources.
These two aspects, chemical processes and groundwater pathways, constitute the entire basis for understanding the limits that chemistry imposes on sustainable yield of aquifers. This chapter considers only aquifers in sedimentary rocks because these are the only significant type in Kansas. Aquifers in crystalline rocks in other parts of the world provide water of different chemical quality, but these are not discussed here. Figure 5.1 shows the general locations of the different aquifers and sites mentioned in this chapter.
Table 5.1 shows the most abundant components of potable ground water, sea water, an average composition for the earth's crust, several important rock types, and the earth's atmosphere. These are grouped as major components and minor or trace components, with an approximate order of magnitude decrease in concentration between categories. The differences among these groups are quite apparent: major chemical elements in the earth's crust are oxygen and silicon, with lesser amounts of aluminum, iron, calcium, sodium, potassium, and magnesium; major dissolved species in sea water are chloride (the ionic form of the element, chlorine), sodium, sulfate, and magnesium. Major components of the earth's atmosphere are nitrogen and oxygen gas. The major-ion chemistry of sea water and the average composition of the earth's crust and atmosphere are stable, for all practical purposes.
Table 5.1--Major and minor components of chemical reservoirs that influence ground-water chemistry.
| Major Components | |||||||
|---|---|---|---|---|---|---|---|
| Atmosphere† | Freshwater | Sea Water‡ | Earth's Crust§ | ||||
| ppm by volume | mg/L | mg/L | elemental, % or ppm |
||||
| N2 | 780,000 | HCO3 | x00 | Cl | 19,350 | O | 46.6% |
| O2 | 209,460 | Ca | x - x00 | Na | 10,760 | Si | 27.72% |
| Ar | 9340 | Na | x - x00 | SO4 | 2710 | Al | 8.13% |
| CO2 | 320 | Mg | x - x0 | Mg | 1290 | Fe | 5.00% |
| H2O | 40-40,000 | SO4 | x - x00 | Ca | 411 | Ca | 3.63% |
| Cl | x - x00 | K | 399 | Na | 2.83% | ||
| K | x - x0 | K | 2.59% | ||||
| Si | x - x0 | Mg | 2.09% | ||||
| Ti | 0.44% | ||||||
| Mn | 0.09% | ||||||
| P | 0.10% | ||||||
| Selected Minor Components | |||||||
| Ne | 18.18 | CO3 | 0.x - x | Br | 67 | Ba | 0.04% |
| He | 5.24 | F | 0.x - x | Sr | 8 | F | 625 ppm |
| CH4 | 1.4 | NO3 | 0.x - x | B | 4.5 | Sr | 375 ppm |
| Kr | 1.14 | Sr | 0.x - x | F | 1.3 | S | 260 ppm |
| H2 | 0.55 | Fe | 0.0x - x | Si | 0.5 - 10 | C | 200 ppm |
| N2O | 0.33 | B | 0.x - X | NO3 | 0.005 - 2 | Zr | 165 ppm |
| Ozone | 0.01-0.03 | Ba | 0.x | Li | 0.18 | V | 135 ppm |
| SO2 | 0.001-0.004 | Br | 0.0x - 0.x | Rb | 0.12 | Cl | 130 ppm |
| I | 0.06 | Cr | 100 ppm | ||||
| Ba | 0.010 | Rb | 90 ppm | ||||
| Mo | 0.010 | Ni | 75 ppm | ||||
| U | 0.0033 | Zn | 70 ppm | ||||
| Cu | 55 ppm | ||||||
† Brownlow, 1996.
‡ Modified from Drever, 1988.
§ From Klein and Hurlbut, 1993.
* "x" indicates order of magnitude; x is 1 to 9 mg/L; 0.x - x means 0.1 to 9 mg/L; x0 means 10 to 90 mg/L; x00 means 100 to 900.
| Major Components | |||||
|---|---|---|---|---|---|
| Basalt | Granite (low Ca type) |
Clays and shales | Sandstones | Carbonate rocks | |
| O | n.g. | n.g. | 52.8% | n.g. | n.g. |
| Si | 23% | 34.70% | 23.8% | 36.80% | 2.40% |
| Al | 7.80% | 7.20% | 10.45% | 2.50% | 0.42% |
| Fe | 8.65% | 1.42% | 3.33% | 0.98% | 0.38% |
| Ca | 7.6% | 0.51% | 2.53% | 3.91% | 30.23% |
| Na | 1.80% | 2.58% | 0.66% | 0.33% | 0.04% |
| K | 0.83% | 4.20% | 2.28% | 1.07% | 0.27% |
| Mg | 4.60% | 0.16% | 1.34% | 0.70% | 4.70% |
| P | 0.11% | 0.06% | 0.077% | 0.017% | 0.04% |
| Ti | 1.38% | 0.12% | 0.45% | 0.15% | 0.04% |
| Mn | 0.15% | 0.039% | 0.067% | 0.00x%** | 0.11% |
| Selected Minor Components | |||||
| Ba | 330 | 840 | 800 | x0 | 10 |
| F | 400 | 850 | 500 | 270 | 330 |
| Sr | 465 | 100 | 450 | 20 | 610 |
| S | 300 | 300 | 3000 | 240 | 1200 |
| C | n.g. | n.g. | 10,000 | n.g. | n.g. |
| Zr | 140 | 175 | 200 | 220 | 19 |
| V | 250 | 44 | 130 | 20 | 20 |
| Cl | 60 | 200 | 160 | 10 | 150 |
| Cr | 170 | 4.1 | 100 | 35 | 11 |
| Rb | 30 | 170 | 200 | 60 | 3 |
| Ni | 130 | 4.5 | 95 | 2 | 20 |
| Zn | 105 | 39 | 80 | 16 | 20 |
| Cu | 87 | 10 | 57 | x ** | 4 |
n.g. not given
* modified from Parker, 1967
** "x" indicates order of magnitude. For example, 0.00x% is 0.001 to 0.009%; x is 1 to 9
Ground-water chemistry, in contrast to earth and seawater chemistry, is strongly influenced by two factors. First, the original depositional water in an aquifer may be flushed out by a chemically different water, and so the degree of flushing determines the chemistry of ground water available to react with the aquifer matrix. The extent of flushing is determined by both the hydraulic properties of the aquifer matrix and climatic conditions during the history of the aquifer. Second, the solid material with which the water has been in contact, and especially the solubilities and reactivities of minerals in the matrix, determine how much the depositional water is modified.
Sedimentary rocks in Kansas were deposited mostly in rivers or areas adjacent to rivers, in deltas which were at the boundary between land and sea, and in shallow-ocean to deep-ocean environments. Thus, the original depositional water for aquifers in Kansas sedimentary rocks ranges from river-like water to sea water. The degree of flushing of an aquifer during a period of relatively unchanging climate depends upon a) the climate (volume of potential recharge water due to the climate), b) the size of the recharge area allowing freshwater to displace depositional water, c) the permeability of the aquifer materials to water flow, and d) to a certain extent, the length of time allowed for flushing. Domenico and Robbins (1985) demonstrated that, after a period of time, a steady-state distribution of water chemistry is reached in a homogeneous aquifer when transport of dissolved ions is accomplished only by fluid flow (advective transport). After a steady state is attained, the redistribution of water chemistry continues but is limited to processes such as diffusion, which is the very slow movement of ions driven by chemical gradients.
For an aquifer with a relatively large recharge area such as the Equus Beds aquifer (fig. 5.1), steady-state ground-water flow is characterized by aquifer fluids similar in composition to the recharge water (as modified by chemical reactions with the aquifer matrix). For an aquifer with a small recharge area (smaller than the breadth of the aquifer), the original depositional water can remain in the aquifer and is distributed so as to envelop the recharging water (fig. 5.2).
Figure 5.1--Location of aquifers and sites in Kansas mentioned in this chapter.
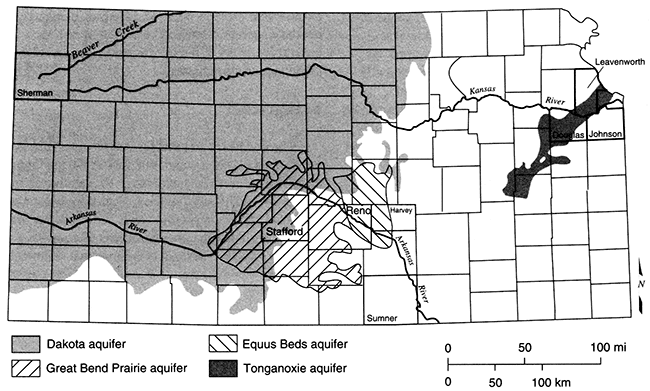
Figure 5.2--Distribution of a conservative chemical species in a homogeneous aquifer after steady-state flow is reached. The width of the recharge area is shown by the hachured area on the y-axis of the plot. Concentration of the dissolved species in the resident fluid was 2,500 mg/L. Contours of concentration of the species show that there is mixing between the resident fluid and recharging fluid (concentration of 10 mg/L); from Domenico and Robbins, 1985.
Original depositional water and recharge water (water displacing or mixing with water present in an aquifer, either naturally or as induced) may have had a distinctive chemistry before contact with the aquifer matrix. Rainwater, as an example of a recharge water, is typically very dilute (low TDS). During storage in the soil zone, however, evapotranspiration (evaporation and water use by plants) usually results in an overall increase in dissolved solids concentration (see Dissolution/Precipitation, below, for more on changes in carbon dioxide content in soil). The increase is not the same for all dissolved components, however: those dissolved components that do not interact with the mineral matrix or biota concentrate in the soil water proportional to the amount of water that evaporates, while those that do interact may increase in concentration but the increase is less than expected from the amount of evaporation. In general, concentration factors of two to ten times are probably typical (Gerritse and Adeney, 1992). Because of differential concentration of dissolved components, ratios of components are frequently used to identify rainwater that has been concentrated in the soil zone. Sea water, an example of a depositional water rather than a recharge water, is saline (high TDS). It contains some components that are stable through time and others that participate in several kinds of geochemical and biochemical reactions. Determination of reliable indicators of remnant sea water continues, as investigators discover previously unsuspected sources or sinks for various elements (Fabryka-Martin et al., 1991; Land, 1995a).
A precursor saline water may be suspected if TDS and especially chloride are present in areas thought to be poorly flushed. The section of the Dakota aquifer (figs. 5.1, 5.24B) in northwestern Kansas, for example, contains saline, chloride-rich ground water. Portions of the Dakota to the south and southeast of the saline area, especially those in the subcrop and outcrop of the Dakota, today contain much fresher water. Present flow directions in the Dakota are from recharge areas in central and southern Colorado to the east and east-northeast (Macfarlane et al., 1989; see also Chapter 4, this volume). In addition, local recharge in the outcrop area in central Kansas (also the regional discharge area) creates local flow cells with much fresher water. The precursor fluid in the freshwater portion of the Dakota cannot be determined directly, but the structural history of the region and the saline region in northwestern Kansas provide clues. Tectonic tilting in Kansas related to the most recent major uplift of the Rocky Mountains has made western and northwestern Kansas topographically higher than it was before that uplift (Merriam, 1963). This elevation change may have been great enough to radically change the ground-water-flow directions in the Dakota, originally from southeast to northwest, to the present direction of west to east. The Dakota saline water in northwestern Kansas may thus be the remnant of the unflushed, down gradient portion of an ancient flow system. That flow system would have exhibited a salinity gradient of freshwater in the southeast and progressively more saline water toward the northwest. Even though this hypothesis is difficult to prove, it suggests that the precursor fluid in the freshwater portion of the Dakota was chemically related to the saline water in northwestern Kansas, and that, because of present-day flow directions, the saline fluid in northwestern Kansas occupies a stagnant portion of the aquifer.
A typical aquifer matrix consists of minerals that are chemically stable to unstable and may also contain varying amounts of organic matter that may be highly reactive to fairly inert. Chemical reactions between water and aquifer matrix change the proportions of dissolved species in the water when relatively long time periods are involved (days to years, depending upon the reaction). Some reactions are facilitated by bacteria and may involve the aquifer matrix or just the water. These reactions generally follow the rules of kinetics, in which there is a reaction rate that is limited by such things as temperature, nutrients, or other conditions in the aquifer.
In chemical reactions, the products of the reactions may contain elements in the same form, even as the same molecule. Alternately, the products of the reactions may contain the elements in a different phase (such as transformation from liquid to solid or liquid to gas) so that the chemical reactions cause removal (or addition) of the element from ground water. In addition, transformations can result from change in redox state (see Boxed section 5.1, Redox) of an element. In these cases, the element is transformed to a different oxidation state (see Boxed section 5.1) which may, in turn, cause transfer of the element to a different molecule. Changes in redox can alter the toxicity of the element or otherwise reduce the usefulness of the water by, for example, causing the water to become corrosive or to deposit scale.
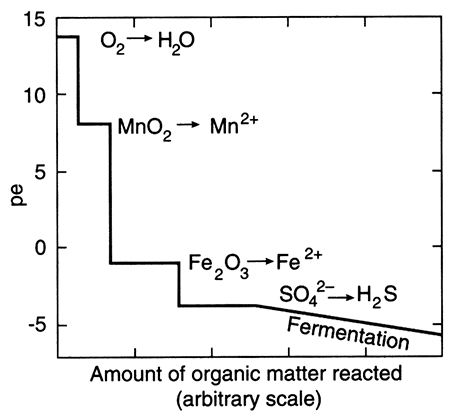
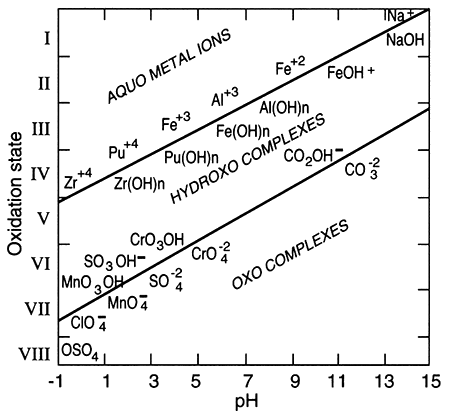
The term "redox" is a contraction of the words reduction and oxidation. The redox state of a fluid represents, in a complex way, whether elements that have multiple oxidation states (and so are redox elements) will tend to be in their higher (more oxidizing) or lower (more reducing) states. The oxidation state of an element is the number of electrons it contains in excess of or as a deficit of the number of protons in its nucleus. Because electrons carry a negative electrical charge and protons a positive electrical charge, the elemental state (protons = electrons) is uncharged. If an element can lose electrons relative to the elemental state, it becomes positively charged (protons > electrons), and if it can gain electrons then it becomes negatively charged (electrons < protons), The oxidation state, then, corresponds to the excess or deficit of electrons. In theory there are steplike changes in the oxidation state of ground water that reflect which species pair is controlling the redox state of the water. The solubility of redox elements often is strongly controlled by pH as well as the redox state, so that the oxidation state of ground water is usually portrayed on a bivariate plot showing pH and a system parameter "pe" or "Eh" that represents the redox state. The parameter "pe" is a calculated measure of the number of electrons and the parameter "Eh" is a measure of the voltage potential that develops between an electrode with a known potential and the solution. As shown below on the figure on the right, uncontaminated ground waters in the world fall within a fairly restricted portion of the total pH-Eh (or pe) region that is theoretically available. Some typical as well as unusual aquatic environments are shown on the figure on the left.
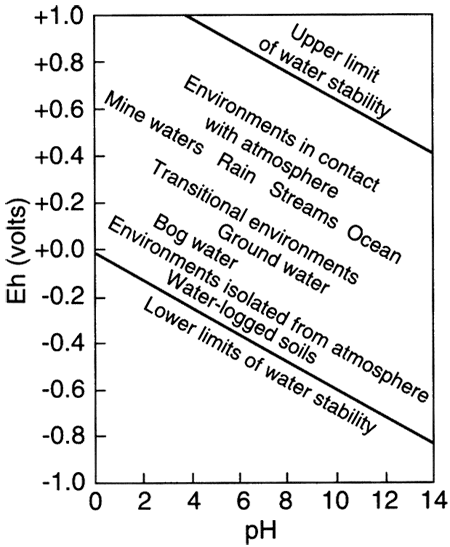
pH-Eh diagram showing generalized positions of some aquatic environments (from Johnson et al., 1989).
Elements with multiple oxidation states that are most important in hydrogeology include oxygen, carbon, nitrogen, manganese, iron, and sulfur.
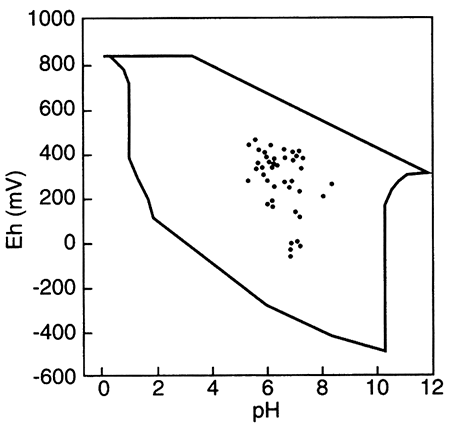
Outlined area shows the region delineated by Bass Becking et al. (1960) for a large number of measurements of natural waters. Small circles are ground-water samples (from Fish, 1993).
The redox state of an aquifer is thought to change in a step-wise fashion with depth or with increasing distance along a flow path. Dissolved oxygen in water yields the highest amounts of usable energy to bacterial mediators as it is reduced to O2, and so is consumed first by oxidation-reduction reactions. Other dissolved substances yield less energy so are used by bacteria next, with consequent change in oxidation state, and so on. Thus, a recharging water, most likely containing dissolved oxygen, is initially highly oxidizing, then makes a step decrease to a lower overall oxidation state as the oxygen is reduced and dissolved oxygen is consumed. Subsequent step-like changes are linked to changes in other metal and nonmetal redox "indicators": the nitrogen system, the iron and manganese systems, the sulfur system, and the carbon-dioxide system. Changes in redox state of a fluid can cause precipitation or dissolution of solids and may result from a) mixing caused by aquifer mismanagement, b) contamination, as well as from c) the normal flushing that occurs when "native" fluids are displaced by different fluids.
(from Drever, 1998; reprinted with permission of Prentice-Hall, Inc.)
The following brief discussion summarizes the terminology used in the rest of this chapter in order to clarify references to these types of transformations. It is important to stress that strict categorization using this terminology is not always possible, as dissolved species that behave one way in one hydrogeologic setting may behave differently in another setting. Therefore, the descriptions used in this chapter for the most part apply to potable ground water.
First, the term conservative is used here to describe a dissolved species with the tendency to remain in the water phase and to not participate in chemical reactions that tend to remove that species from ground water. Conservative elements can be useful as indicators of how the water component of ground water is changing (evaporating, being diluted or mixed with another water). These elements also move at the same speed as ground water, acting as a useful tag of ground-water-flow rate. Nonconservative describes those species that are readily removed from or added to ground water during reaction with a solid phase such as minerals or organic matter. A change in a nonconservative species concentration along a ground-water-flow path implies that a chemical or biochemical reaction between water and the aquifer matrix has occurred.
Labile species are those that change form, either by changing oxidation state or by changing the type of compound in which they are found; this includes transformation from dissolved or liquid state to gaseous state. Refractory species (sometimes known as recalcitrant when only microbially mediated reactions are considered; Johnson et al., 1989) do not undergo these types of transformations. (In this chapter, labile and refractory apply to both inorganic and organic species.) Thus, labile transformations are of two basic types, one involving change in redox state and the second in change from liquid to gaseous state. In summary, a labile species changes forms readily; a refractory species does not. For the most part labile species are nonconservative, although refractory species are not necessarily conservative. Table 5.2 lists the major dissolved species in ground water and some of the minor dissolved species, along with their classifications according to the labile-refractory and conservative-nonconservative terminology. Also included in this table are the Environmental Protection Agency drinking-water standards for the species listed.
Table 5.2--Persistence of dissolved species in water; organics section modified from Domenico and Schwartz, 1990, p. 428; reprinted by permission from John Wiley & Sons, Inc.
| Major Components | |||||
|---|---|---|---|---|---|
| Labile | Refractory | Conservative | Nonconservative | MCL**, mg/l | |
| Cations | |||||
| calcium | Ca | ||||
| magnesium | Mg | ||||
| sodium | Na | Na | tbc* | ||
| potassium | K | K | |||
| Anions | |||||
| bicarbonate | HCO3 | HCO3 | |||
| sulfate | (SO4) | (SO4) | (SO4) | (SO4) | 500 |
| chloride | Cl | Cl | 250† | ||
| Others | |||||
| dissolved silica | H4SiO4 | H4SiO4 | |||
| Selected Minor Components | |||||
| Labile | Refractory | Conservative | Nonconservative | MCL**, mg/l | |
| Inorganic and Nutrients | |||||
| aluminum | Al | Al | 0.05 -0.2† | ||
| arsenic | As | As | 0.05 | ||
| antimony | Sb | Sb | 0.006 | ||
| barium | Ba | Ba | 2 | ||
| beryllium | Be | Be | 0.004 | ||
| bromide | Br | Br | |||
| cadmium | Cd | Cd | 0.005 | ||
| chromium | Cr | Cr | 0.1 | ||
| copper | Cu | Cu | 1.3†† | ||
| cyanide | CN | CN | 0.2 | ||
| fluoride | F | F | 4 | ||
| iron | Fe | Fe | 0.3† | ||
| lead | Pb | Pb | 0.015†† | ||
| manganese | Mn | Mn | 0.05† | ||
| mercury | Hg | Hg | 0.002 | ||
| nickel | Ni | Ni | 0.14 | ||
| ammonium as N | NH4-N | NH4-N | |||
| nitrate as N | NO3-N | NO3-N | 10 | ||
| nitrite as N | NO2-N | NO2-N | 1 | ||
| phosphate as P | HPO4-P | HPO4-P | |||
| selenium | Se | Se | 0.05 | ||
| silver | Ag | Ag | 0.1† | ||
| thallium | Tl | Tl | 0.002 | ||
| vanadium | V | V | |||
| zinc | Zn | Zn | 5† | ||
| Organics | |||||
| Labile | Refractory | Conservative | Nonconservative | MCL**, mg/l | |
| Volatile Organics | |||||
| trichloroethylene | TCE | TCE | 0.005 | ||
| benzene | C6H6 | C6H6 | 0.005 | ||
| toluene | C6H5CH3 | C6H5CH3 | 1 | ||
| xylenes (total) | e.g., 1,2-(CH3)2C6H4 | xylenes | 10 | ||
| vinyl chloride | CH2=CHCl | CH2=CHCl | 0.002 | ||
| carbon tetrachloride | CCl4 | CCl4 | 0.005 | ||
| Pesticides & Herbicides | |||||
| Atrazine | Atrazine | Atrazine | 0.003 | ||
| Chlordane | Chlordane | Chlordane | 0.002 | ||
| Dioxin | Dioxin | Dioxin | 3 x 10-8 | ||
| 2,4-D | 2,4-D | 2,4-D | 0.070 | ||
| Heptachlor | Heptachlor | Heptachlor | 0.0004 | ||
**EPA Maximum Contaminant Level, in mg/L. Complete listing of regulated chemicals with maximum contaminant levels, maximum contaminant-level goals, secondary maximum-contaminant level, and toxicity doses can be found on the EPA Web page: http://water.epa.gov/drink/contaminants/index.cfm
†: secondary maximum contaminant level
††: action level; standard is "zero"
*tbc: to be considered
Naturally occurring dissolved species in ground water are present because of: a) rainwater recharging an aquifer, b) degree of concentration of rainwater in the soil zone before moving into an aquifer, c) presence of depositional water, though probably altered by biogeochemical reactions, d) chemical reactions between water and soil, including organic matter, e) chemical reactions between water and rock, and f) biological action.
Ground-water chemistry in carbonate (limestone, dolomite) aquifers is relatively simple because of the relatively simple mineralogy in the aquifer. Ground water in these aquifers is typically a calcium bicarbonate type water, with varying amounts of magnesium and sulfate, depending on the amounts of dolomite and gypsum or anhydrite, respectively, present (e.g., White, 1988).
Progressive, normal changes in ground-water chemistry because of water-rock interaction in siliciclastic aquifers (sandstone, sand and gravel, or similar aquifers; fig. 5.3) result in a continuum from calcium bicarbonate dominated water through sodium bicarbonate, sodium sulfate, and sodium chloride dominated water when freshwater progressively displaces a sodium chloride type water such as sea water. These changes reflect the importance of dissolution of calcite (calcium bicarbonate water), cation exchange on clay minerals (sodium bicarbonate water), and finally an interface with the less-flushed part of the aquifer (sodium sulfate and sodium chloride type waters). Two examples of deviations from this normal progression in water chemistry are found in the Dakota aquifer of Kansas.
Figure 5.3--Typical changes in ground-water chemistry with time and distance along a flow path result in changes in major dissolved ions. Shown are artificial data simulating the typical evolution; arrows show direction of movement. This type of presentation is known as a Piper diagram (Piper, 1944), in which triangles show proportions of cations and anions, and data from the triangles are projected onto the central diamond along lines parallel to one side of the triangles. Circles indicate TDS, and in the typical progression, ground water increases in TDS as it changes from a calcium-bicarbonate water (Ca-HCO3) near a recharge area to a sodium-chloride water (Na-Cl) near the farthest extent of flushing of the aquifer.
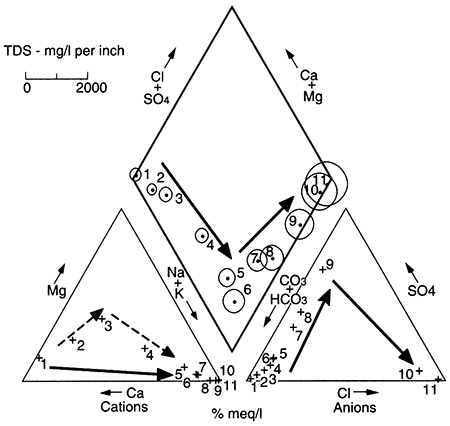
The Dakota aquifer (fig. 5.1) crops out at high elevations in Colorado, where it is recharged. Ground water travels generally eastward through the subsurface in eastern Colorado and western Kansas, and then discharges in the vicinity of the outcrop and subcrop of the Dakota in central Kansas (see Chapter 4, this volume). Chemistry of the Dakota ground water in the Kansas outcrop area resembles recharge-area ground water, because the outcrop is being recharged by local meteoric water as well as by ground water that has travelled through the Dakota from Colorado and western Kansas (Macfarlane et al., 1989). Besides the situation just described in the Dakota aquifer in Kansas, the case in which there is a very long flushing time or the case in which there is no precursor saline fluid in an aquifer also results in the absence of the sodium chloride and possibly sodium sulfate type waters at the end of the flow path.
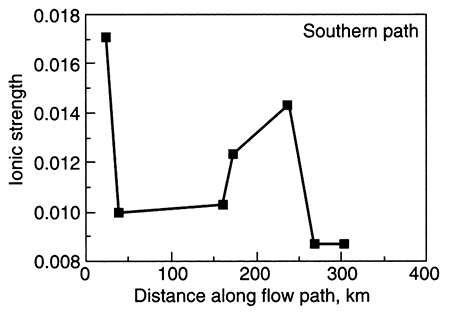
A second example showing a variance from normal ground-water chemistry changes occurs when there is cross formational leakage of a fluid into the central part of an aquifer. Leakage of, for example, a saline, sodium chloride type water into an aquifer at the midpoint of the flow system results in a sodium chloride facies present at the aquifer midpoint. The other water types develop on either side of the point of leakage. The Dakota aquifer in Kansas shows such a pattern of water-chemistry types (fig. 5.4 and fig. 5.24 later in this chapter; Macfarlane et al., 1989). In general, the rate of leakage, water quality, and volume of leakage collectively determine the seriousness of this phenomenon. In the case of the Dakota aquifer, the change from a chloride content of less than 100 mg/L to more than 500 mg/L occurs in the vicinity of cross formational leakage of sodium chloride (Na-Cl) brines from Permian aquifers underlying the Dakota. The increase in TDS limits the usefulness of Dakota ground water in west-central Kansas.
Figure 5.4--Change in ionic strength along a ground-water flow path (A-A' shown in fig 4.5) in the Dakota aquifer, Kansas. Ionic strength is a representation of the concentration of dissolved ions in a solution and is directly proportional to total dissolved solids. Notice the abrupt increase in ionic strength in the middle part of the flow path. This is the locus of cross formational flow of deeper, saline water into the Dakota.
Contaminants introduced into ground water are affected by many of the same processes as natural recharge: a) mixing with in situ water, b) evaporative concentration (usually restricted to the soil zone), c) reaction with the aquifer matrix, and d) biological alteration. The discussion below elaborates on these processes in terms of conservative versus nonconservative species and labile versus refractory species. Chemical and biochemical reactions are discussed in terms of their reactivity instead of according to the distinction between chemical and biological, to focus on how water chemistry can change and thus be a factor in determination of safe yield.
Ground water may gain or lose dissolved species as it moves through the soil zone and aquifer, when it mixes with chemically different water, or merely as a matter of time when chemical reactions are slow. The following sections describe the most abundant dissolved chemical species in ground water and how likely they are to be involved in chemical reactions. Although not comprehensive, the sections focus on the most abundant dissolved species or the most important chemical reactions. Also included is a brief discussion of the implications to the mixing of chemically different ground water, and contrasts of "fast-path" ground-water flow (found in fractured rocks) and "slow-path" ground-water flow (found in most other circumstances).
Conservative dissolved species in ground water, those not easily removed from solution, may fingerprint a single source of that particular dissolved species. In practice, there are often several potential sources for these species, so that identification of the contributing source(s) becomes difficult to impossible. For some elements the use of isotopic ratios (see Boxed section 5.2, Isotopes and Ground Water; Glossary, isotope) provides a better fingerprinting tool than the element itself. In addition, some isotopes decay spontaneously to other elements at a steady rate, making them useful for dating the water and, of course, making them nonconservative. The use of isotopes in studying ground-water chemistry is explained in many textbooks (e.g., Ferronsky et al., 1982; Bowen, 1988; Drever, 1988; Faure, 1986; Pearson et al., 1991) and will not be discussed further here.
Isotopes of the same element contain the same number of protons but different numbers of neutrons. Isotopes are radioactive (parent) if they decay spontaneously to another (daughter) element. Isotopes are stable if they do not decay. Most elements in the periodic table have one or more naturally occurring stable isotopes and some have a radioactive isotope.
Radioactive isotopes can be useful in dating some aspect of ground water, because radioactive elements decay spontaneously and constantly, the constancy permitting the time calculation. The decay time is often expressed in terms of the half-life, the amount of time needed for one-half of the starting material to spontaneously decay to the daughter product. To calculate the age, one solves for "t" in the following equation:
Pt = Po e-kt
where P denotes parent isotope,
t denotes condition at some time,
o denotes condition at the initial time,
k is the decay constant; the half-life, T0.5, is related to k according to: T0.5 = ln(2)/k.
The half-lives of several radioactive isotopes are listed below, to demonstrate the possibility of dating the time of ground-water recharge (time of removal from the atmosphere) over quite a long time. These are not the only radioactive isotopes used for dating in hydrogeology.
| Radioactive Isotope |
Half Life | Useful Dating Range |
Source† | |
|---|---|---|---|---|
| Tritium | 3H1 | 12.3 yrs | ≈100 yrs from 1954 |
2 |
| Argon | 39Ar18 | 270 yrs | 50-1,000 yrs | 1 |
| Silicon | 32Si14 | 276 yrs | 50-1,500 yrs | 1 |
| Carbon | 14C6 | 5,730 yrs | 1,000-75,000 yrs | 1,2 |
| Chlorine | 36Cl17 | 301,000 yrs | 80,000-1,500,000 | 1,2 |
| † Principal source for radioactive isotopes: 1 cosmic bombardment of the earth's atmosphere, incorporated in rain, snow, etc. 2 atmospheric testing of weapons, 1952 to 1969; incorporated in rain, snow, etc. |
||||
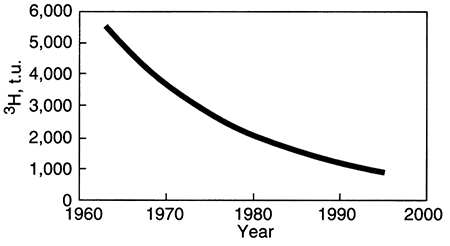
Although most sources of radioactive isotopes are natural, a few were been artificially increased during restricted periods in the recent past. Tritium and carbon-14 were both byproducts of atmospheric testing of bombs during the 1950's and 1960's. Therefore, ground water that was recharged during that time contained an unusually high amount of tritium. The graph below illustrates the theoretical amount of tritium (3H1) in such a ground water that was recharged from atomospheric precipitation in 1963. Radioactive decay causes a decrease in concentration through time, but if that ground water were sampled in 1997, it would contain much more tritium than today's rain contains (10 to 20 tritium units, t.u.), thus identifying it as water recharged during the era of atmospheric testing of weapons. This is an example of a dating technique that is useful in a general way in that it identifies a period of time during which the ground water might have been recharged. Other radioactive isotopes can allow more precise dating of the time of recharge.
As shown below, water recharged in 1963 might have contained about 5,500 tritium units (t.u.). By 1997 (the end of the trend line), the amount of tritium left in such a water is about 800 t.u., much higher than modem rainwater. Because the amount of tritium in rainwater during the 1950's and 1960's was not constant, the exact age of a ground water with high tritium levels cannot be uniquely determined, but high tritium levels (in the absence of contamination from nuclear reactor cooling water, for example) indicate recharge during the era of atmospheric bomb testing.
Stable isotopes provide the opportunity for a different kind of interpretation of chemical processes. Stable isotopes do not transform, but because there is a small difference in mass of the element because of the different number of neutrons in the nucleus, chemical or physical processes that discriminate for or against higher mass can alter an isotope ratio: the ratio of two isotopes of the same element. The larger the relative change in mass between the two isotopes, the greater the discrimination, or fractionation. Listed following are isotope ratios commonly used to look for fractionation processes in ground water.
| Element | Isotopes | Relative mass difference |
Processes that fractionate |
|
|---|---|---|---|---|
| Hydrogen | 2H1 | 1H1 | 2/1:100% | evaporation of water, formation of clay minerals, temperature of recharge water |
| Oxygen | 18O8 | 16O8 | 18/16:12.5% | evaporation of water, formation of clay minerals, temperature of recharge water |
| Carbon | 13C6 | 12C6 | 13/12:8.33% | biological processes, CO2 solution/exsolution, oxidation/reduction of C (organic matter; CO2 based species) |
| Nitrogen | 15N7 | 14N7 | 15/14:7.1% | oxidation/reduction of N (in organic matter; in gases or of nitrate) |
| Sulfur | 34S16 | 32S16 | 34/32:6.25% | ocean evaporation (ratio preserved in gypsum), oxidation/reduction of S (pyrite; gypsum) |
Stable isotope ratios are compared to a standard and expressed according to the following notation.
δR in units of ‰ (per mil) = [(Rsmpl - Rstd) / Rstd] * 1000
where
R is the ratio of the low-abundance to the high-abundance isotope,
n is the mass number of the low-abundance isotope,
smpl is the unknown sample, and
std is the international standard.
The graph below shows the meteoric water line, a relationship between δD (or δ2H) and δ18O that is nearly contant in rainwater around the globe.
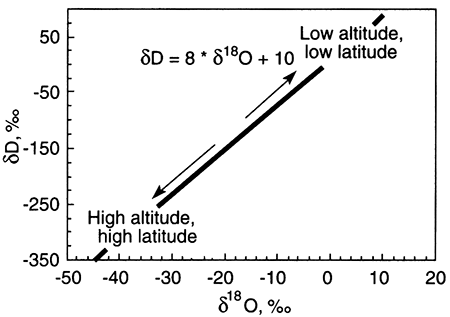
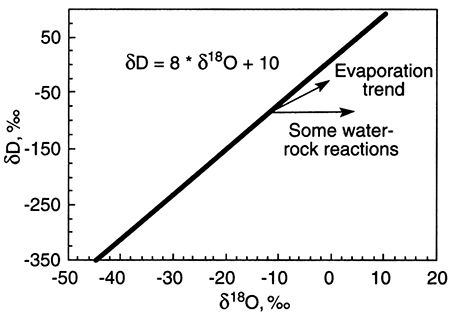
Ground water with δD and δ18O values that do not fall on the meteoric water line has undergone alteration from rainwater. Alteration processes include evaporation before recharge of the rainwater to ground water and water-rock interaction. The trends from the meteoric water line that these processes take are shown on the plot below. The starting point for any alteration process is the δD-δ18O composition of rainwater that recharges ground water.
More information about stable and radioactive isotopes can be found in Ferronsky and Polyakov (1982), Bowen (1988), Drever (1988), Faure (1986), and Pearson et al. (1991), as well as in other similar books.
The single dissolved species in potable ground water that is considered a major component and that is conservative is chloride (Cl-). The two sources for naturally occurring chloride in ground water (excluding chloride resident in the depositional water) are chloride found in rainwater that recharges ground water and chloride added to ground water when it dissolves halite (NaCl). Halite may originate from dry fallout (dust carried by wind and deposited on vegetation and the land surface) or from large salt deposits such as those found in salt domes or bedded halite. In the latter case, chloride concentrations in ground water can be very high near the halite source and decrease away from it, making it easily identified as a point source. In temperate to humid climates where large salt deposits are absent, halite should be extremely rare in aquifers because it is highly soluble and thus should be quickly dissolved from an aquifer. Nevertheless, halite is often declared the source of chloride in water when chloride content cannot be attributed to rainfall recharge. In these cases, dry fallout, precursor saline fluid resident in low-permeability areas of an aquifer, or cross formational flow of saline water may be the source.
Sulfate (SO4-2) is typically conservative but labile (can change form) under highly reducing conditions (see Labile and Refractory Species section following). When sulfate concentration in ground water is higher than in soil water, minerals are the source of the sulfate. (Soil-water sulfate may be higher than rainwater sulfate because of evapotranspiration.) Under oxidizing conditions, sulfate can be added to ground water through transformation from the sulfide form of sulfur (such as pyrite or other mineral sulfides). Mineral sulfides may supply a significant amount of sulfate to ground water, especially in water that is in contact with coal or lignite (Hem, 1985). Ground water with moderate to high concentrations of sulfate, however, probably acquired the sulfate from other minerals or from evapoconcentration processes. Gypsum (CaSO4·2H2O) and anhydrite (CaSO4) are naturally occurring minerals with solubilities that are high, but not as high as halite. In Kansas, gypsum can form in the soil zone when evapotranspiration rates greatly exceed precipitation, so that water is evaporated from the soil leaving the salts behind (Moran et al., 1978). Gypsum is one of the salts commonly observed in dry soils in temperate climate regions (Sposito, 1989) and in arid regions, white efflorescence at the surface is often a sodium-sulfate precipitate (Hem, 1985). Wetting and drying cycles through the seasons can lead to pulses of sulfate recharging an aquifer: evapoconcentrated soil solutions are flushed into the aquifer during a heavy rain after a dry spell, or gypsum precipitated in the soil can be redissolved in the rainwater moving down through a soil. The high solubility of gypsum guarantees that persistence is unlikely during wet periods. Because very dry conditions are necessary for precipitation of gypsum, dissolved sulfate in ground water is very nearly a conservative species.
Most other major dissolved species in ground water, calcium (Ca+2), magnesium (Mg+2), sodium (Na+1), potassium (K+1), and bicarbonate and carbonate (HCO3-1 and CO3-2) are nonconservative. They participate in one or more common chemical reactions that either remove them from or add them to ground water, as discussed in the next section.
Three categories of chemical reactions involving nonconservative species are presented below. Dissolution and precipitation of solids affects concentrations of major dissolved species and some minor species in ground water. Sorption of dissolved ions onto surfaces of solids exerts a strong control on the concentration of minor dissolved species in ground water. Ion exchange, in which ions move in and out of selected locations within a mineral structure, strongly affects the major-ion content of a ground water and may also affect the minor-ion content.
Water dissolves all solids to some extent. The most soluble minerals (those which result in the most mass in a unit volume of water) are generally the least available for dissolution because they are so soluble (Domenico and Schwartz, 1990). The most soluble minerals, the most common being halite and gypsum, generally only provide dissolved ions to water and do not precipitate from ground water (an exception being gypsum in the soil zone, as discussed above). Excluding the most soluble common minerals, then, it can be said generally that small adjustments in ground-water chemistry occur constantly as the water both dissolves and precipitates solids in order to be in a state of equilibrium with them all at the temperature and pressure in the aquifer.
Commonly occurring minerals that dissolve congruently (completely, with no reaction products being solids) and that have an important influence on ground-water chemistry are calcite, dolomite, and quartz. In restricted regions, evaporite minerals such as halite and gypsum can completely control the chemistry of ground water, but these cases will not be included here because of the focus on potable ground water.
Quartz is the principal mineral in most sandstones and so is quite abundant. Dissolution of quartz (SiO2) results in the addition of dissolved silica, known as silicic acid (H4SiO4), to water, although it is not the sole source. (Note that silicic acid does not behave as an acid in typical ground waters, that is, it does not yield its hydrogen ions to attack other minerals.) This reaction (simplified, as are all others in the chapter) may be represented by
SiO2 + H2O ↔ H4SiO4 (eq. 5.1)
quartz + water ↔ silicic acid
The dissolution of quartz provides a component of dissolved solids to ground water that is useful to interpreting origins of water chemistry. For example, silicic-acid content can be used to differentiate end-member waters in a mixture (D. O. Whittemore, personal communication, 1994) and can be used to calculate the in situ temperature of the ground water as a geothermometer, making use of the well-characterized dependence of solubility of quartz on temperature (Siever, 1962; Fournier and Potter, 1982).
The amount of calcite and dolomite that will dissolve in water depends upon the partial pressure of carbon dioxide (P-CO2) in the water. The acid created by dissolving CO2 in water attacks these two minerals. The dissolution of CO2 is expressed as:
CO2 + H2O ↔ H2CO3 (eq. 5.2)
carbon dioxide + water ↔ carbonic acid
The acid, H2CO3, is called carbonic acid, and again the chemical reaction is written with a double arrow to make it clear that the reaction can go in either direction: if there is more CO2 in the "atmosphere" in contact with the water than can be dissolved in the water, then H2CO3 is created as the reaction proceeds to the right and the "atmosphere" is robbed of CO2 and water. If conditions change (e.g., water exits the ground to form a spring, or something acts to consume the CO2 in the "atmosphere," or the temperature of the water increases as the water moves deeper into the ground), then the reaction proceeds to the left, consuming H2CO3 and producing CO2 and water. Carbon dioxide is present in fairly low concentrations and thus the P-CO2 is low in the atmosphere (table 5.1), resulting in fairly low concentrations in rainwater. In soils, however, the activity of bacteria increases the amount of CO2 by a factor of 10 to 100 or more, providing most of the CO2 for water-rock reactions.
Calcite and dolomite are important contributors to the chemistry of potable ground water in that they are relatively easily dissolved and are fairly abundant in rocks. Calcite releases calcium ions and bicarbonate ions to water during dissolution:
CaCO3 + H2CO3 ↔ Ca+2 + 2 HCO3-1 (eq. 5.3)
calcite + carbonic acid ↔ calcium + bicarbonate
Dolomite dissolves in a similar fashion:
CaMg(CO3)2 + 2H2CO3 ↔ Ca+2 + Mg+2 + 4 HCO3-1 (eq. 5.4)
dolomite + carbonic acid ↔ calcium + magnesium + bicarbonate
Calcite is found almost everywhere. Dolomite also is very common and is frequently cited as the principal source for Mg+2. Almost all calcite contains small amounts of Mg+2 in the mineral lattice, as well, and almost certainly provides a significant amount of Mg+2 to ground water. In addition, recent work suggests ion exchange to be a very important contributor of Mg+2 (Appleo, 1994; Chu, 1995; see Ion Exchange, following).
A precipitate from ground water is known as an authigenic mineral (one formed in place, not transported) if it was precipitated some time after the aquifer was deposited and as cement if it acts to bind loose sediment together. Thus, both the grains (sediment) and cements or authigenic minerals can dissolve to change water chemistry. Furthermore, there may be several generations of cement, recording a history of the fluids and the temperature-pressure conditions to which the sediment has been exposed. The most common cements in sandstones are calcite and quartz; the most common cement in limestones is calcite. Dissolution/precipitation reactions of these minerals were covered above.
Major dissolved species that are nonconservative in ground water, then, include calcium, magnesium, and bicarbonate, because they are part of the reactions dissolving and precipitating calcite and dolomite. Bicarbonate can also be added to water during the weathering of other rock-forming minerals, especially the feldspars but also other alumino-silicates. Quartz, a major component of sandstones, is slightly soluble and its dissolved counterpart, silicic acid, also is nonconservative.
Sorption is an equilibrium reaction just as dissolution/ precipitation reactions are. Sorption describes the affinity of a dissolved species for the surface of a solid. The solid phase involved in sorption can be mineral, which strongly attracts many dissolved metals, or organic, which strongly attracts many hydrophobic organic compounds as well as metals.
Three groups of mineral-like substances, aluminum, iron, and manganese oxyhydroxides, are inorganic substances that are strong sorbers (Drever, 1988). These substances are present in most sandstone aquifers and may also be present in limestone aquifers. They include many different forms of oxides and hydroxides, many of which are hydrous (contain water as part of their structure). In addition, they are highly reactive and often exist as coatings on other grains. Because the grain size is very small, the surface area of these substances is large (often on the order of 200 m2/gm: Drever, 1988), and thus their absorptive capacity is large. Most mineral surfaces sorb numerous kinds of dissolved ions due to the electrostatic attraction between the ion and the surface and because of complexation reactions that occur at the surface (Johnson et al., 1989), but the aluminum, iron, and manganese oxyhydroxides are the strongest sorbers among minerals or mineral-like substances (or, more generally, inorganically formed solids).
Solid organic material in aquifers sometimes has a large sorptive capacity (van Riemsdijk and Hiemstra, 1993), although the complexity of organic compounds and their sorption behavior makes it difficult to quantify organic sorption (Drever, 1988). Organic material varies in reactivity according to its type and maturity: it may come from terrestrial matter (plant debris, mostly) or marine organisms (in ancient rocks) and changes as it ages, both in texture and in composition (e.g., Dow, 1978; Waples, 1980; Magoon and Dow, 1994). Humus is the dark-colored solid matter in soil that includes all organic components except for those identifiable as unaltered or partially altered biomass; humus comprises humic substances and biomolecules (Sposito, 1989). The humic substances that persist in the soil profile are the most studied fraction of organic matter in soils. These substances are depleted in nitrogen and enriched in sulfur relative to soil organic matter as a whole. The much higher carbon-to-nitrogen (C/N) ratio of humic substances is an indication of the change from material highly susceptible to microbial attack to material resistant to attack (Sposito, 1989).
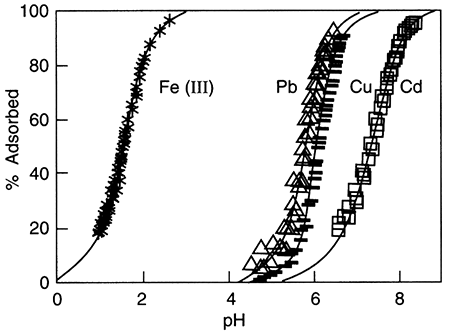
Sorption of metals from water onto inorganic surfaces is often pH-controlled. The pH of the water (see Glossary, pH) controls the attachment of H+ or OH- ions to surfaces, which in turn causes them to have a positive or negative charge (Stumm and Morgan, 1981). Sorption of ferric iron (FeIII) onto a hydrous oxide surface removes from about 20% to almost 100% of the iron in solution as the pH varies from 1 to 3. For the same surface, lead and copper sorption from solution ranges from less than 5% to about 90% over the pH range of 4.5 to 6.5, and cadmium sorption ranges from about 20% to about 95% over the pH range of about 6.5 to 8.5 (fig. 5.5). In these cases, there is always less sorption at lower pH than at high pH, because of the charge on the metal ion. Negatively charged ions behave in the opposite way, being more strongly sorbed at lower pH than higher. It may also be true that chemical forces in addition to electrical forces, are involved in sorption processes. Murray (1975) demonstrated that significant sorption occurred at the zero point of charge on manganese oxide (δ-MnO2), the point at which the surface is neither negatively nor positively charged.
Figure 5.5--Metal-ion adsorption on amorphous silica as a function of pH (Johnson et al., 1989). For each metal shown, adsorbed amount decreases with decreasing pH. Furthermore, Fe(III) is completely sorbed at pH of about 3, while lead, copper, and cadmium are still dissolved.
Organic matter also has a pH-dependent, negative surface charge (Riemskijk and Hiemstra, 1993). Metals that commonly sorb onto organic matter include aluminum (Al), vanadium (V), chromium (Cr), manganese (Mn), iron (Fe), nickel (Ni), copper (Cu), zinc (Zn), cadmium (Cd), and lead (Pb) (Sposito, 1989). Organic matter provides an important limit to concentrations of potentially toxic metals in ground water.
Sorption affects the concentration of dissolved organic compounds as well as dissolved metals in ground water. Two different phenomenon create the potential for sorption. First, organic molecules in aqueous solution are attracted by the net negative surface charge on humus (solid organic matter). Second, sorption occurs when electrostatic forces binding dissolved organic molecules to organic solids are stronger than the forces holding organic molecules in water (Sposito, 1989). The distribution of organic molecules between water and organic matter is an equilibrium process and the amount sorbed becomes strongly a function of the amount of organic matter present. Organic compounds that react with soil organics in rural areas come mostly from agricultural chemicals and their degradation products. In urban areas, organic compounds that can react with soil organics include lawn-care products, oil and grease, and household chemicals. Soil humus may effectively arrest movement of these compounds into the subsurface. However, some compounds may become complexed with dissolved organic compounds, facilitating their movement into and through aquifers. A general rule is that the more soluble in water an organic compound is, the smaller is its tendency to sorb onto organic (or other) substances (Sposito, 1989).
In summary, sorption is a reversible process that describes the affinity of dissolved species for the surfaces of solid materials, either organic or inorganic. Because the process is reversible, metals or organics sorbed to surfaces can be des orbed if aqueous solution composition changes. This creates the possibility for addition of species to water through desorption that were not a component of the displacing aqueous solution (fig. 5.6). The mixing of fluids that can result from poor aquifer management could conceivably create such a situation.
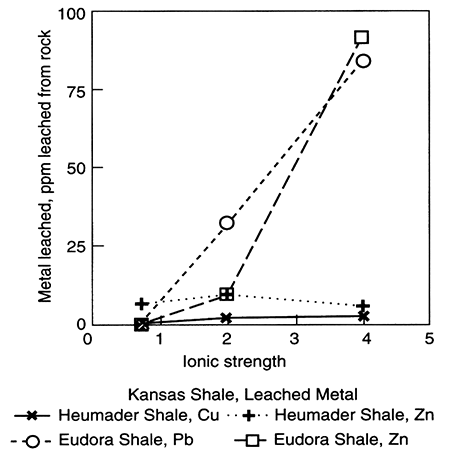
Figure 5.6--Leaching shales with various salinities of Na-Cl type water causes metals such as copper (Cu), zinc (Zn), and lead (Pb) to leave sorption or ion-exchange sites and reside in the leaching water. This effect depends upon salinity (here reported as ionic strength) and also temperature and type of salt. Note that movement of saline fluids from deeper aquifers (see section III) could cause metals to leach from sediments; data from Long and Angino (1982). Both shales are from Kansas.
An important mechanism of nonconservative behavior of ions in solution is ion exchange. The process of ion exchange requires a so1id phase that has a charge deficiency (is negatively charged) for exchange of cations, or a charge excess (is positively charged) for exchange of anions. Most solids that are important ion exchangers affect cations in solution, although anion exchange can occur in some circumstances (Drever, 1988, p. 218-219).
Clay minerals are important ion exchangers that acquire their charge imbalance because of imperfect chemical composition, most often because of substitution of an aluminum ion (+3 charge) for a silicon ion (+4 charge). The clay-mineral lattice then acquires a permanent negative charge that is not affected by the pH of the surrounding solution. The negative charge attracts cations into specific sites within the layered structure of the clay mineral. The layers containing the exchangeable ions also contain water molecules, thus providing relatively easy ingress and egress for the exchanging ions.
Capacity for exchange is determined empirically, by measuring the uptake and release of ammonium ions from a sediment sample. This Cation Exchange Capacity (CEC) is frequently reported in units of milliequivalents per 100 grams (meq/100 gms), and is the amount of electrical equivalents of ions divided by 1,000 per 100 gms of sediment. Different clay minerals have different ion exchange capacities. The highest CEC of the common clay minerals is on the order of 80 to 150 meq/100 gms (smectite or montmorillonite clays). Another group of clay minerals has intermediate to low CEC at 10 to 40 meq/100 gms (illite clay minerals), and other clay minerals have very low CEC at less than 10 meq/100 gms (kaolinite and chlorite clays; Drever, 1988).
Ion exchange is an equilibrium process, in which the exchange of one ion for another onto a solid material is described by a constant. As with other equilibrium processes, the constant represents the ratio of the products of the chemical reaction to the reactants. A typical ion exchange reaction is:
Na-solid + K+1 ↔ K-solid + Na+1 (eq. 5.5)
(sodium on the exchange sites of a solid) + (dissolved potassium) ↔ (potassium on the exchange sites of a solid) + (dissolved sodium)
This exchange is one atom of sodium for one atom of potassium and is driven by equilibrium processes. A more complicated situation exists for exchange of ions that have difference valences:
Na2-solid + Ca+2 ↔ Ca-solid + 2Na+1 (eq. 5.6)
(sodium on the exchange sites of a solid) + (dissolved calcium) ↔ (calcium on the exchange sites of a solid) + (dissolved sodium)
This exchange is two-for-one in order to satisfy the electrical charges in the solution. Constants derived from experiments producing ion exchange are highly sediment and ion specific and are difficult to generalize. It is well established, however, that solids exhibit a preference for ions that is charge- and size-dependent. The order of cation affinity for exchange is known as the Hofmeister series (Stumm and Morgan, 1981), and is written from left to right in order of decreasing preference of the solid for the ion (and so increasing likelihood of the ion residing in the solution):
Cs+1 > K+1 > Na+1 > Li+1
for the common monovalent ions
Ba+2 > Sr+2 > Ca+2 > Mg+2
for the common divalent ions
← preferred on the solid preferred in the liquid →
Describing preference order for a combined monovalent and divalent list is more difficult because the exchange is strongly controlled by the total concentrations involved (e.g., Drever, 1988). In essence, when estimating exchange between Ca+2 and Na+1, Ca+2 is the preferred ion on the solid phase when TDS content is low, and Na+1 is the preferred ion on the solid phase when TDS content is high, even if the ratio of Na+1 to Ca+2 content stays the same. A generalized list of ion exchange affinities (at approximately the same total concentrations) could be as follows (Domenico and Schwartz, 1990), again in order of most strongly preferred on the left to least preferred on the right:
Al+3 > Ca+2 > Mg+2 > NH4+1 > K+1 > H+1 > Na+1 > Li+1
← preferred on the solid preferred in the liquid →
The process of ion exchange is completely reversible and occurs relatively rapidly as long as water (and dissolved ions) can pass freely through the sediment. An important caveat is that clay minerals, the best ion exchangers, make up the largest portion of low-permeability materials (mud layers and shales). The equilibration of low-permeability material with water recharging adjacent high-permeability materials is slow simply because of the time it takes the recharging water to penetrate the low-permeability material. Thus, mud or shale provides a reservoir of exchangeable ions that can influence the chemistry of water within an aquifer over a long period of time.
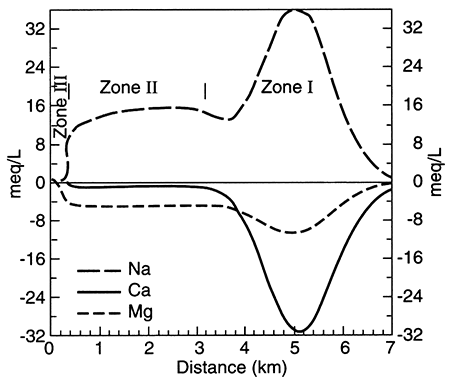
Recent work from two independent studies demonstrates the long time required for equilibration of marine shales with recharging (fresher-than-seawater) water (Appelo, 1994; Chu, 1995). In Kansas, the Dakota aquifer (fig. 5.1) is a series of sandstones and mudstones deposited in fluvial (river) and deltaic settings. Chu (1995) used a computer model to simulate the patterns observed in the water chemistry of the Dakota aquifer. Cation-exchange processes, thought to be the most important type of reaction affecting the water chemistry in the Dakota, are apparently ongoing. The computer model showed that, using reasonable numerical values to represent the Dakota's chemical properties, equilibration of clay minerals with the freshwater recharging it is not yet complete, even though the marine or marginal-marine clays were deposited more than 65 million years ago (fig. 5.7). In the Dakota sandstones, the chemical patterns seen on maps of the Dakota water chemistry evolve through time as incoming freshwater displaces resident water and ion exchange of different ions occurs (Chu, 1995). The best approximation of the evolution of ion exchange is that calcium and magnesium first replace sodium on ion-exchange sites and later calcium replaces magnesium on ion-exchange sites. The lateral (map-view) sequence created (fig. 5.8), from the downgradient saline fluid to the upgradient, fresher-water part, is a) a saline (Na-Cl) fluid-Zone I, b) a Na-HCO3 type fluid created as ion exchange processes are more or less complete-Zone II, c) a Na-Mg type fluid and a Mg type fluid that is usually transient and not observed-Zone II-Zone III transition, and d) a Ca-HCO3 or Ca-Mg-HCO3 fluid that represents recharge water in steady-state equilibrium with the aquifer matrix-Zone III. This sequence indicates that the clays are still yielding ions from ion-exchange sites that were saturated with sodium when the clays were in contact with sea water (see table 5.1 for the composition of sea water).
Figure 5.7--The ratio of (Ca+Mg)1/2 / (Na) in units of mols, versus TDS for field data (points) and a computer simulation (line). Sea-water composition is shown for comparison (*). The identified zones represent different stages of completion of ion exchange in the Dakota aquifer; from Chu, 1995.
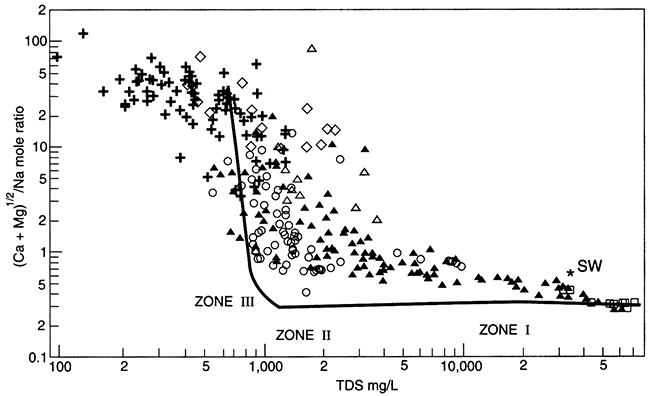
Figure 5.8--Modelled concentrations of sodium, calcium, and magnesium in the Dakota Aquifer along a groundwater-flow path. The plot shows concentrations in excess of (positive values) or as a deficit from (negative values) concentrations expected from mixing with no chemical reaction of resident fluid and recharging fluid. The pattern shows expected changes in water chemistry because of cation exchange; from Chu (1995).
In summary, the conservative and nonconservative dissolved species discussed above usually make up the largest proportion of dissolved solids in potable ground water. The chemical behavior of labile and refractory species, discussed in the next section, is important. This is because the transformation that they may undergo can cause them to be removed from ground water, either through precipitation as a solid or through volatilization, and/or the transformation can render them either less or more harmful than before the transformation.
Labile species typically found in ground water include those that undergo changes in redox potential and those that are volatile. Refractory species are those that mayor may not have the capacity to undergo transformations but do not undergo transformation under ordinary groundwater conditions at significant rates. The following discussion focuses on labile species but is limited to those most commonly studied in potable ground water.
Two fundamental kinds of transformations are discussed in the following sections, those in which organic compounds are transformed into other compounds and those in which there is a change in redox state of an element. Transformation processes often facilitate removal of a compound from water through precipitation in a solid phase or through change into a gas phase. Of course, the redox change can work in the opposite direction, to add components to water. Both types of transformations are typically mediated by bacteria, meaning that conditions in the aquifer must be favorable for activity of the appropriate bacteria. Pumping a well can alter conditions in the aquifer, potentially either inducing or inhibiting these transformations. The change occurs principally because of introduction and mixing of different kinds of water from above, below, or laterally within the aquifer (see below, The Consequences of Mixing).
Transformations of organic compounds occur because of hydrolysis (reaction with water to attach OH-l or H+l to the compound, usually making it more water soluble), ionization (stripping off a H+l, making the compound more water soluble), and biodegradation (a general term describing all reactions in which bacteria transform compounds; Johnson et al., 1989). In addition, organic compounds vary tremendously in their solubility (how much dissolves in water) and volatility (tendency to form a gas).
Redox reactions are those that involve the transfer of electrons and thus are involved in oxidation (loss of electrons) or reduction (gain of electrons), resulting in a change in valence for the species. Some elements can lose Or gain as many as eight electrons and can exist in multiple oxidation states with different valences. Elements behave differently depending upon oxidation state, in that they exist in different kinds of solids (with different solubilities) and molecules and affect animals differently upon ingestion. The redox state of a fluid (see Boxed section 5.1, Redox) is a complex representation of the oxidation states of all the redox elements present in a fluid.
For this discussion of labile species in ground water, the focus is on a nutrient (nitrate), organic compounds, and metals with multiple redox states. Although there are others, these are discussed because they are the most common contaminants in ground water and most often limit its use. Introduction of one or more of these species because of poor management of an aquifer could render the ground water unacceptable for its intended use, and thus these species have the most impact on issues of safe yield.
Nitrate is one of the most common contaminants of ground water. It is harmful in very high concentrations to livestock and in only moderately low concentrations to human infants (EPA maximum contaminant level is 10 mg/L nitrate-nitrogen. Nitrate concentration can be reported as nitrate ion [NO3-1], with a drinking-water limit of 45 mg/L, or as nitrate-nitrogen, with a drinking-water limit of 10 mg/L). Nitrate almost always enters aquifers from the land surface, and thus factors that accelerate downward movement of water can in turn accelerate entrance of nitrate to an aquifer. Nitrate mayor may not persist in water, as discussed below.
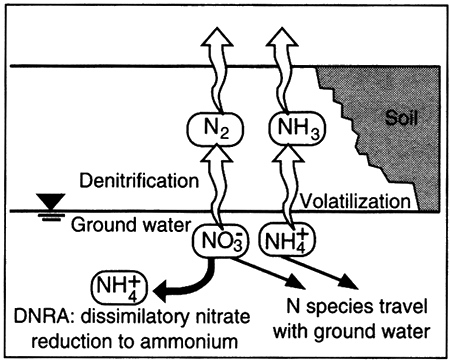
Dissolved nitrogen in ground water occurs in many forms, from the most oxidized form, nitrate (NO3-1), to the most reduced form, ammonium (NH4+1). Transformations among nitrogen species are typically mediated by specific genera of bacteria and are often studied within the context of the nitrogen cycle (see Boxed section 5.3, The Nitrogen Cycle).
The ultimate source of nitrate is the atmosphere, which is mostly nitrogen gas (N2). Although bacteria transform small amounts of N2 to NO3-1 in root nodules on legumes, most naturally occurring nitrate comes from decay of organic material that contains small amounts of nitrogen relative to carbon, hydrogen, and oxygen (Drever, 1988):
C106H263O110N16P + 138 O2 -> 106 CO2 + 16 NO3-1 + HPO4-2 + 122 H2O + 18 H+1 (eq. 5.7)
where C106H263O110N16P = organic matter,
CO2 = carbon dioxide,
NO3-1 = nitrate,
and HPO4-2 = phosphate
Large quantities of nitrate are introduced to ground water through human activities. Oxidation of animal waste to nitrate (barnyards, feedlots, septic systems) and nitrogen-based fertilizers in agricultural regions are the two most important sources of introduced nitrate in ground water.
The two principal pathways by which nitrate is reduced are 1) through nitrous oxide species to nitrogen gas (N2; called denitrification) and through nitrite (NO2-1 to NH4-1 (called DNRA, dissimilatory nitrate reduction to ammonium; Korom, 1992; Smith et al.; 1991; Postma et al., 1991). Denitrification can be accomplished by bacteria using organic carbon as a source for electrons to reduce the nitrate (heterotrophic bacteria) or by using another source of electrons (autotrophic bacteria), as shown by the following two equations:
4NO3-1 + 5C + 2H2O → 2N2 + 4HCO3-1 + CO2 (eq. 5.8)
where 5C + 2H2O = organic matter, simplified representation
6NO3-1 + 2FeS2 + 2H2O → 3N2 + 2FeOOH + 4SO4-2 + 2H+1 (eq. 5.9)
where FeS2 = pyrite,
and 2FeOOH = goethite
DNRA proceeds generally from nitrate to ammonium, and may occur where nitrate amounts are limited:
NO3-1 → NO2-1 → NH4+1 (eq.S.lO)
Requirements for these reactions are the presence of nitrate-reducing bacteria (e.g., Thiobacillus denitrificans is one bacteria that denitrifies), oxygen-limited conditions in the aquifer (dissolved oxygen content less than about 2 mg/L; Hendry et al., 1983), and the other nutrients necessary for the bacteria. Absolute controls on rates of denitrification are not well established (Korom, 1992), but an excess amount of easily oxidized material in an aquifer (such as plant debris) can reduce even high supply rates of nitrate to nitrogen gases and prevent contamination of an aquifer (Simpkins and Parkin, 1993).
Poor aquifer management can create a nitrate problem in ground water. Much of fertilizer nitrate is used by plants or denitrified in the soil zone. Rapid passage of recharge water through the soil zone and bypass of the soil zone through flow in fractures can reduce the effectiveness of plant use, denitrification, and DNRA in minimizing nitrate content of recharge water. For those systems in which DNRA dominates, introduction of oxygen-rich water into a reducing environment can reverse the DNRA reaction, effecting transformation of NH4+1 to NO3-1 and thus creating a nitrate problem. Issues associated with vertical movement of ground water and lateral movement as induced by pumping are discussed later in this chapter (see Inter-aquifer Ground-Water Flow).
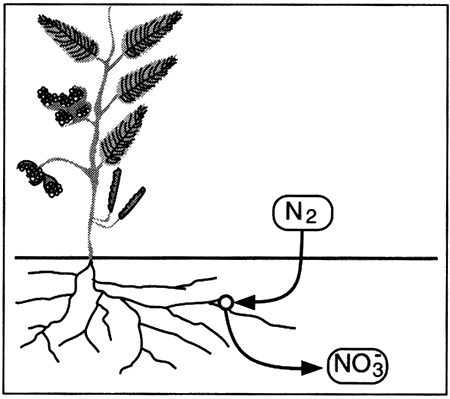
Nutrients such as nitrogen are cycled through biological materials, the atmosphere, soils, and water in a continuous loop. The cycle, in a natural world, completely accounts for all available nitrogen, and none of the various parts of the cycle gain or lose nitrogen except as changes in climate affect the transformation processes. In the natural cycle, rain and snow (precipitation) containing nitrate and ammonium fall on the earth.
If the rain soaks into the soil to become part of the soil water, the nitrate and ammonium ...
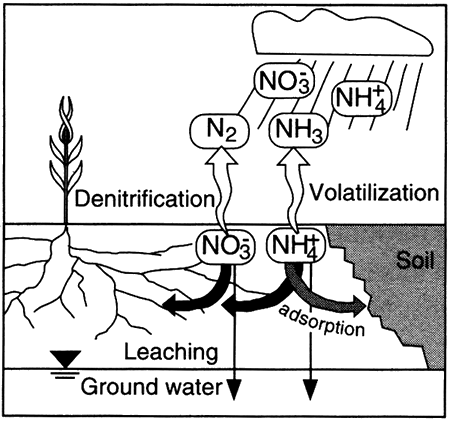
The nitrate and ammonium in the plants ...
Ammonium that is adsorbed ...
Nitrate and/or ammonium in the ground water ...
Nitrogen in the atmosphere ...
Other sources of nitrogen, from human activities, are fertilizer and animal waste such as sewage, barnyard waste, and feedlot waste. Nitrogen from these sources is subject to the natural processes described above, but overloading of nitrogen in an area can result in natural processes unable to transform the nitrogen species as quickly as they are supplied, with the result that ground water typically becomes enriched in nitrate.
Example: Equus beds, south-central Kansas--The Equus beds aquifer in south-central Kansas (fig. 5.1) is the principal source of irrigation, domestic, and municipal water in that region. Unconsolidated Pliocene and Pleistocene sand, gravel, and silt deposits comprise the high-permeability but heterogeneous aquifer. Ground water in the Equus beds in Harvey County has been studied in some detail. Harvey County has little topographic relief except for sand dunes in restricted areas. The thickness of the unsaturated zone and the saturated thickness of the aquifer thins from west to east across the county. Total dissolved solids in the ground water vary between about 250 and 1,000 mg/L, with major dissolved components being calcium, sodium, bicarbonate, and sulfate.
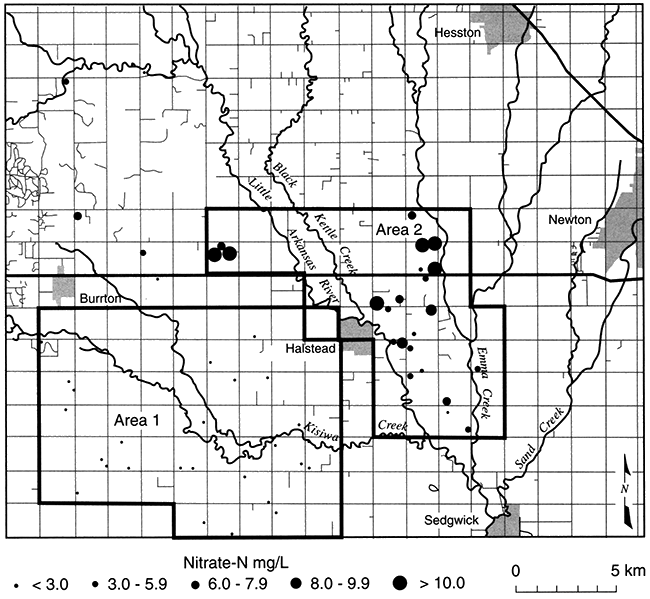
Crops are irrigated and fertilized in Harvey County, making conditions favorable for nitrate contamination of ground water, but measured NO3-N concentrations are low to undetectable in part of the county (area 1, fig. 5.9; Townsend and Sleezer, 1994 and 1995). Nearby (area 2, fig. 5.9), even though dryland farming techniques are used, NO3-N concentrations are up to three times the EPA Maximum Contaminant Level (MCL). This suggests that conditions are favorable for denitrification in area 1 but not in area 2. A reducing environment in area 1 is indicated by detectable dissolved iron (Fe+2) and ammonium, and a slight hydrogen sulfide (H2S) odor. Nitrogen isotope ratios of dissolved nitrate show enrichment of the 15N isotope: the pairing of low-nitrate concentrations with enriched 15N is a conventional indicator of nitrate reduction. In addition, sulfate concentrations are higher than in nearby areas, which, in conjunction with dissolved iron, suggests that iron sulfides may be the electron donor to nitrate reduction. Townsend and Sleezer (1995) showed that an average of 9-20 mg/kg of NO3-N was stored in the upper 3 m (10 ft) of the vadose zone within the selected field-site region. Thus, nitrate is available for leaching between the land surface and 3-m (10-ft) depth. High concentrations of nitrate in this upper soil zone contrast with decreased concentrations in ground water beneath, suggesting that the conditions for denitrification are met somewhere between 3- and 10-m (10-33-ft) depth.
Figure 5.9--Nitrate-N concentrations in domestic wells, Harvey County, Kansas (Townsend and Sleezer, 1995).
Therefore, in Harvey County, conditions in the Equus beds aquifer and the overlying unsaturated zone are optimum for the reduction of nitrate in some places, and in other places not, even in the same general area. Understanding what controls denitrification is an ongoing area of research, and teasing out the sensitivity of an aquifer to contamination remains an important goal.
Organic molecules contain reduced carbon (-IV to 0 valence) in combination with other elements, dominantly hydrogen and oxygen. The structure of organic molecules can be quite complex, but generally the more complex an organic molecule the less likely it will dissolve in water and the more likely it will remain as a separate phase (table 5.3). Organics as a separate phase can be more dense than water (dense nonaqueous phase liquids, DNAPL's) or less dense than water (light, nonaqueous phase liquids, LNAPL's) and consequently move downward in an aquifer by gravitational forces or float on water, respectively.
Table 5.3--Solubility of selected organic compounds. [More complex molecules generally have a higher molecular mass, and are thus less soluble; modified from Domenico and Schwartz, 1990; reprinted by permission from John Wiley & Sons, Inc.]
| Compound | Molecular Mass (g/mol) | Solubility (g/m3) |
|---|---|---|
| C6H6 (Benzene) | 78.0 | 1780 |
| C7H8 (Toluene) | 92.0 | 515 |
| C8H10 (o-Xylene) | 106.0 | 175 |
| C9H12 (Cumene) | 120.0 | 50 |
| C10H8 (Naphthalene) | 128.0 | 33 |
| C12H10 (Biphenyl) | 154.0 | 7.48 |
In addition to size of the molecule, a few other general rules control how well organic molecules dissolve in water. First, organic compounds may be classified as polar or nonpolar, depending upon whether the electrical charge of the compound is distributed unevenly (in two poles) or evenly around the molecule. Because water is a polar molecule, it dissolves polar organic molecules much more readily than nonpolar ones. Second, in general, organic matter is the primary material for sorption of organic molecules, so the more organic material in the host rock of an aquifer the more organic sorptive capacity of the aquifer. Finally, chemical reactions involving organic molecules are usually kinetic reactions: the rate of chemical reaction often depends upon the concentration of one or more participants in the reaction, and the rate of reaction is slow enough that it can be measured.
The EPA list of priority pollutants, with established maximum contaminant levels, is about 75% organic compounds. Research has focused on the breakdown pathways of these components. Most of the early data on the various degradation processes of the organic priority pollutants are summarized in Mabey et al. (1982). Principal degradation processes and controls on solubility of organic molecules include molecule size and charge, volatility, hydrolysis, ionization, and biodegradation (e.g., Johnson et al., 1989).
In general, whether aquifer management accelerates movement of organic compounds depends on the properties of the organics. The tendency to be removed from solution increases if the organic compound has low solubility and is volatile, is easily sorbed, or can be easily hydrolyzed or ionized and thus made more susceptible to biodegradation. Biodegradation, if complete, reduces organic compounds to harmless CO2 and water; if incomplete, it can, in some cases, create compounds more harmful than their precursors.
Some metals lose or gain electrons as a way of improving stability in solutions (Stumm and Morgan, 1981, p. 323). The transition from one redox state to another for an individual element is often pH as well as pe dependent (see Boxed section 5.1, Redox; fig. 5.10). Changes in redox state often cause dissolution or precipitation of metal oxides, hydroxides, and sulfides, and indirectly cause precipitation or dissolution of carbonates or other minerals. The redox state of an aquifer system (see Boxed section 5.1, Redox), as indicated by the pe, is buffered by the rocks or sediments that contain the water. Thus, the redox state is not expected to change significantly unless there is a radical change in water moving through the aquifer (see Consequences of Mixing, p. 149) or the aquifer is composed of unreactive minerals (such as very pure quartz sand), making it poorly buffered.
Figure 5.10--Generalized depiction of the stability areas for some redox-sensitive elements (from Stumm and Morgan, 1981; reprinted by permission of John Wiley & Sons, Inc.).
Several metals are affected by the redox state of water (table 5.4). Some general statements can be made about the effect of a change in redox state on the mobilization of metals. Because elements with multiple redox states are usually also affected by pH, both factors are considered in the groupings found in table 5.4. The table is not comprehensive, but lists a few elements of interest in ground water and the conditions under which they are mobile. Some elements or groups of elements are mobile under more than one set of pH-redox conditions. The different sets are numbered.
Table 5.4--pH-redox stability regions for selected ions. [pH and redox conditions given show the general ranges for stability of the species in solution (in the dissolved form).]
| Element(s) | pH Conditions | Redox Conditions |
|---|---|---|
| As (arsenic) | 1) moderate to low | 1) reducing |
| 2) high | 2) all | |
| Hg (mercury) | 1) all | 1) oxidizing |
| Cu (copper) | 1) low | 1) oxidizing |
| 2) high | 2) intermediate | |
| Fe (iron), Mn (manganese), Pb (lead) | 1) low | 1) all |
| Ni (nickel), Co (cobalt), U (uranium) | 1) low | 1) all |
| 2) high | 2) intermediate to oxidizing | |
| Cr (chromium) | 1) all but intermediate | 1) intermediate to reducing |
Under other pH and redox conditions, the above metals are immobile. By effecting a change from a condition under which a metal is immobile to one in which it is mobile, dramatic changes in water quality can occur.
Redox zonation exists naturally in ground water but also can be created by introduction of foreign materials such as landfill leachate. Baedecker and Back (1979) demonstrated that the compressed chemical zonation of an aquifer in the vicinity of a leaking landfill is similar to the broader zonation that occurs in marine sediments (fig. 5.11). Near the landfill where leachate is "fresh," the reduced forms of carbon, nitrogen, and sulfur (methane, ammonia, and hydrogen sulfide) exist in preference to the oxidized forms. Iron and manganese are found in solution in abundance. Farther from the landfill, the oxidized forms of carbon, nitrogen, and sulfur (dissolved CO2 species, nitrate, and sulfate) become dominant in the leachate plume and the presence of oxygen in the water makes dissolved iron and manganese precipitate as oxyhydroxides (more on this topic in a later section, under Contamination, Landfills).
Figure 5.11--Chemical zonation caused by landfill leachate leaking into an aquifer (from Baedecker and Back, 1979). Ground water is reducing near the landfill (contains methane, CH4, hydrogen sulfide species, HS-, and ammonium, NH4+) and is progressively more oxidizing (contains carbon dioxide species like bicarbonate, HCO3-, sulfate, SO4+2, and nitrate, NO3-) as it mixes and reacts with resident, oxidizing ground water.
In summary, many metals (and some nonmetals) are stable in different oxidation states under normal groundwater conditions, and transformation among oxidation states can affect the metals' mobilities. Many transition metals are mobile under acidic, reducing conditions, but some may also be mobile under very alkaline conditions as well. Some metals are immobile under reducing conditions and mobile under oxidizing conditions. In many cases the situation is complicated by the fact that mobility is affected not only by pH and redox conditions, but also by the presence or absence of other phases or dissolved species, such as minerals that can sorb the metals under certain redox conditions (Matisoff et al., 1982) or dissolved species such as sulfides that precipitate fairly insoluble solids with the transition metals.
Ground water generally moves vertically downward through the unsaturated zone to the water table. Unsaturated flow occurs where pore spaces in the soil profile are not completely filled with water. In the unsaturated zone, physical and chemical interactions occur among air, water, and sediment. Much of aquifer recharge occurs from the land surface through the unsaturated zone. Flow through the unsaturated zone is complicated by the presence of rapid flow paths called macropores (such as large pores, burrows and channels, roots, fractures) and by restrictions to flow, exemplified by discontinuous silt or clay layers. The source of the recharge water, the rapidity with which the recharge reaches the aquifer, and the chemical reactions that occur in the unsaturated zone all determine the quality of the water recharging an aquifer.
Examples of the types of water that can enter the unsaturated zone include rainfall, surface water during overland flow or flooding, and playas and other temporary holding ponds for surface water. Sources of recharge water that may contain undesirable chemicals or undesirable concentrations of salts include point sources of liquids such as landfills that produce leachate, spills at agrichemical preparation locations on farmsteads, and irrigation water. The unsaturated zone is composed of the root zone (upper 1.2 m [4 ft]); the intermediate vadose zone, which is the zone between the root zone and the capillary fringe above the water table; and the capillary fringe (fig. 5.12). The capillary fringe is the zone where air in the soil zone meets a fluctuating water table. The thickness of the capillary fringe depends upon the soil-grain size, finer-grained sediments allowing thicker capillary fringes to form. The elevation of the top of the capillary fringe fluctuates with the amount of recharge, just as the water table fluctuates.
Figure 5.12--Schematic diagram showing a cross sectional view of the unsaturated zone and upper part of an aquifer.
Movement of water through the soil profile is complex because of the variety of forces that affect the flow process. The major impact on flow in the unsaturated zone is the effect of the surface area of soil particles and small soil pores that adsorb water (fig. 5.13). Particles with large surface area can adsorb water more tightly than particles with small surface area. Small soil pores hold water more tightly than large soil pores because of capillary attractive forces. Therefore, water drains through large pores more easily than through small pores and, by extension, sandy soils with large pore spaces will drain more easily than soils composed of clays or mixtures of clay, silt, and sand.
Figure 5.13--Cross section of a soil pore. Water is held more tightly near soil particle surfaces (hatchured areas). At some distance away water is weakly held and available for drainage by gravity (from Miller and Donahue, 1990).
In general, water flows from areas of high-water potential to those of low-water potential. The gradient created by an area of high-water potential forces flows from wetter to drier soils or from areas of high pressure to those of low pressure. In most situations the direction of flow is from the land surface downward, although situations occur where capillarity and evapotranspiration cause water to migrate upward through the soil profile toward land surface.
The two end-member types of unsaturated flow are diffuse flow and macropore flow. Diffuse flow is the slow movement of a wetting front through a porous media (e.g., soil and unconsolidated sediments) to the ground-water table. In diffuse flow, water moves slowly from pore to pore displacing air and/or water in its path. Diffuse flow represents a relatively smooth migration of water through interconnected pores.
Macropore or preferential flow involves the rapid movement of water down a vertically interconnected pore network or fracture. Macropores commonly are caused by cracks in the soil due to drying, physical structure of the soil, decayed root channels, abandoned worm tubes, or chemical changes within the soil itself. Macropores can permit rapid and massive movement of water (with contaminants, if present) to the ground-water table without the benefit of retardation or filtration by the soil. Macropore flow can result in contaminants arriving at the water table faster than expected. Tracer tests, designed to determine flow rates in the unsaturated or saturated zones, can be used to identify macropore flow by comparing expected and observed arrival times of a tracer chemical: a tracer that arrives more quickly than expected from the permeability of the soil indicates macropore flow.
The rapidity of transport into aquifers through the unsaturated zone strongly influences some chemical processes. Thus, scenarios predicted for ground water in porous media, where water moves relatively slowly, are different from scenarios predicted for ground water in fractured rocks, where water can move rapidly through open or nearly open fractures. In addition, an aquifer that is composed of porous media, whether consolidated or unconsolidated, will have a moderate to thick unsaturated zone overlying it and aquifers composed of fractured media often have very thin unsaturated zones overlying the aquifer proper. Fractures are nearly always present in limestones and other carbonate rocks, may be present in consolidated sandstones and shales, and may also be present in thick deposits of very fine grained material such as glacial till. Fractures are unlikely in unconsolidated material composed primarily of sand or gravel.
Aquifers present in Kansas are of both types: porous media such as unconsolidated sediment (examples being river alluvium, the Ogallala/High Plains aquifer, and the Great Bend Prairie aquifer) or sandstone (such as the Dakota aquifer), and fractured media (principally limestone, such as the Cottonwood Limestone of east-central Kansas and the Cambrian-Ordovician or Ozark aquifer system of southeastern Kansas).
Soil stratigraphy, describing nonuniform, heterogeneous soil, is one factor that affects flow in the unsaturated zone. Soil is typically composed of multiple layers of sands, silts, clays, caliche, and possibly mineralized zones in a heterogeneous mix. Water movement is strongly affected by the stratigraphy of and structure within the soil and, in particular, relatively large effects are caused by discrete, impermeable layers. The presence of impermeable clay or clay/silt layers in the unsaturated zone can result in a perched water table above the layers (fig 5.14a), retarded flow around or through the clay layer (fig. 5.14b), or flow downgradient with the clay zone acting as a major diversion point (fig. 5.14c; Miller and Donahue, 1990; Kung, 1990). Finer-scale effects occur where there are gradations in soil-grain size and texture. These gradations enhance entrapment of air that in turn affects permeability to water. As stated above, flow is generally from areas of wetter soil to dryer soil (high-water potential to low-water potential), and variable soil stratigraphy increases the magnitude of wet-to-dry gradients. The latter phenomenon results from the relation between permeability and moisture content in unsaturated sediments: maximum permeability occurs in fully saturated sediments, and permeability decreases nonlinearly with decrease in moisture content (Domenico and Schwartz, 1990).
Figure 5.14--Perched aquifers. a) Occurrence of perched water-table aquifer because of permeability differences between fine-textured zones and surrounding sediment. b) Flow is retarded because of permeability differences. In addition to perching, water can flow around fine-textured zones and also eventually saturate the zone (small arrows). c) Slight changes in dip of strata can permit down gradient movement of water as it is deflected by less-permeable zones.
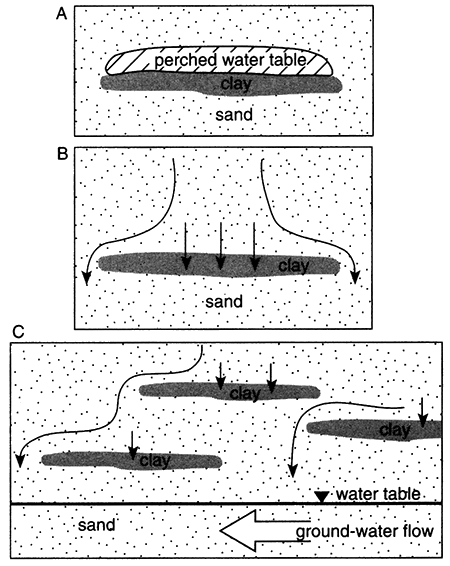
Unsaturated flow is generally vertical in coarse-textured soils such as loamy sands, more lateral in fine-textured soils such as clay loam, and inhibited when a sand occurs below a fine-textured soil because of a "shadow" effect (fig. 5.15; Miller and Donahue, 1990). Fine-textured soil lenses can create a perched water table if the water supply is large enough (fig. 5.14a). With or without a perched water table, flow will not occur through a sand underlying the fine-textured layer, even though the sand is inherently more permeable when wet, until the fine-textured unit is saturated.
Figure 5.15--Typical wetting patterns expected for saturated and unsaturated flow as influenced by texture and location of the water source. Saturated soil-water flow, which always has unsaturated flow occurring at the same time, is mostly vertical in coarse-textured soils (loamy sand), more lateral in fine-textured soils (clay loam). A temporary inhibition to flow occurs when sand is below a finer-textured soil (clay loam over sand; from Miller and Donahue, 1990).
The slow movement of water through the unsaturated zone has implications for safe-yield policies. In areas where extensive pumping occurs, depletion of aquifers surrounded by semi-confining layers that limit recharge can result in the total loss of a usable aquifer. In addition, continuous withdrawal of water can cause the collapse of fine-textured layers. This subsurface collapse can propagate to the land surface, creating large areas of subsidence that may be accompanied by small faults. Land subsidence because of ground-water pumpage exceeding recharge rates has been observed in numerous places, including Houston, Texas (up to 2.7-m [8.9-ft] land-surface subsidence); Mexico City, Mexico (up to 8 m [26.4 ft]); Venice, Italy (up to 3 m [9.9 ft]); and Long Beach, California (up to 9 m [29.7 ft]; Pipkin, 1994). The geology of these regions where subsidence occurred is characterized by relatively thick, fine-grained, unconsolidated, water-saturated sediment (clay and silt) confining an aquifer. The reduction in pore pressure that occurs as water is removed from the underlying aquifer causes compaction of the overlying, fine-grained material. The collapse is not reversible, so areas that have subsided will not "rise" again when pumping stops. This might occur in only a few locations in Kansas. Potential areas of subsidence in Kansas include floodplains of large rivers where muds are deposited over sands and gravels or parts of the glacial-drift aquifer.
The presence of clay/silt layers also causes interactions with the dissolved ions carried by water. Three aspects of this interaction are briefly discussed here. First, clays act as adsorption points for anions and cations, the magnitude of which depends upon the type of clay and the pH of the water. The attraction of anions or cations to a clay surface causes a decrease of ion concentration in the water or change the pH of the water, which in turn may permit further dissolution of other minerals in the soil as the water moves downward. Adsorption of ions to clays can also disperse the clays, thereby decreasing the permeability of the soil. Decreases in the quantity of recharge that reaches the ground-water table have a direct impact on the safe-yield development of an aquifer.
Second, clays can act as agents of cation exchange, where cations of different sizes and valences occupy interlayer sites within the clay-mineral structure. The most common type of cation exchange occurs when water moving through a soil is a calcium-bicarbonate type water and the clays are sodium-rich. Equilibrium between the water and the clay is approached as calcium exchanges for (trades places with) sodium in the clay mineral. Calcium is preferred on the cation-exchange sites in clays in dilute water because calcium is smaller and has a larger charge (+2 versus + 1 for sodium) than sodium. Conversely, when sodium-rich water is used for irrigation, the increase of sodium in the soil zone can increase the dispersive properties of the soil zone, decrease the permeability, and increase the sodium concentration in water that moves downward through the soil zone. These effects are detrimental to the soil both from a chemical and tilth point of view.
A third way in which clay layers can affect the movement of water is by acting as a barrier to flow, thereby creating a local perched aquifer. The water in the perched aquifer may allow chemical reactions that are not possible in the rest of the unsaturated zone. For example, if the temperature and organic carbon content are suitable and anaerobic bacteria are present, denitrification (see previous section, Nitrate) can occur in the perched aquifer, resulting in a decreased nitrate content and increased bicarbonate content. In addition, ponding can permit more rapid evaporation of water in the surface layers of the soil resulting in a zone of increased salinity. Such zones can cause a soil to become sodic. The increased salinization over time will degrade both the soil and the water moving through the soil zone to the ground water. This will be discussed further in the next section.
Caliche zones in the unsaturated zone result from the effects of an arid climate on the movement of water through the soil profile. Rainwater has low total dissolved solids and a low pH, meaning that it is aggressive in terms of dissolving minerals in the soil. Furthermore. there is more carbon dioxide in the soil zone than in the earth's atmosphere because of the breakdown of organic matter and root respiration. The partial pressure of carbon dioxide is a measure of the amount of carbon dioxide in the air: for example, the total soil-atmosphere pressure is the sum of the partial pressures of the individual gases in the soil atmosphere, such as N2, O2, CO2, and much smaller amounts of other gases. The higher partial pressure of CO2 in the soil atmosphere affects the recharging water by lowering the pH of the water. Calcite (calcium carbonate) dissolution results from this more acidic water, causing the pH to rise. In arid and semi-arid regions, the deeper part of the soil profile has less biological activity and the resulting partial pressure of carbon dioxide is lower. Recharge water carried downward through the high carbon dioxide zone to the lower carbon dioxide zone becomes oversaturated with respect to calcite and so it precipitates. Furthermore, evaporation in the upper part of the soil zone concentrates the dissolved solids and draws water back toward the land surface. Both the evaporation of the water and the upward movement cause more precipitation of calcite through a number of complicated steps (Buol et al., 1989).
Caliche zones are commonly found throughout soil profiles in semi-arid areas such as western Kansas but are also routinely found at the base of finer-textured horizons in areas with fluctuating water tables, such as in south-central Kansas. Like clay/silt horizons, caliche zones can act as perching zones or as points of diversion as water migrates through the soil profile to the ground water. In addition, because calcite dissolution causes pH to become more basic. the change may affect other pH-sensitive chemical reactions in the soil, such as volatilization of ammonia gas (NH3(g); see Boxed section 5.3, The Nitrogen Cycle, and related text above). The volatilization of ammonia reduces the concentration of the pool of nitrogen available for nitrification, thus causing a reduction in nitrate in the system. The change in pH may also affect precipitation, transformation, or mobility of other contaminants.
Sources of ground-water recharge include precipitation (both rainfall and snowmelt), irrigation, and leakage from other sources such as playas, landfills, storage tanks, ponds, farmsteads where agrichemicals are handled, and factories. Recharge can also occur vertically upward from underlying aquifers.
Precipitation provides some of the most chemically active recharge in the hydrologic system. Precipitation, unaffected by industrial sources of atmospheric pollution, has a pH of about 5.6 (Hem, 1985) and is very poorly buffered (pH is easily changed). The average pH of precipitation from 1983 to 1991 at one site in east-central Kansas was 5.1 ± 0.6; in southeastern Kansas, pH of rainfall averaged 4.88 and in western Kansas, pH of rainfall averaged 5.83 for water year 1992-93 (Geiger et al., 1994). Variations in pH are due to the sources of ions in the water, the effects of dust moving through the atmosphere and interacting with precipitation, and the effects of surface-soil conditions when rainfall occurs (Randtke, 1996, personal communication). The presence of high P-CO2 in the soil atmosphere (see equation 5.2, above, and related discussion) greatly increases the acidity of the recharging rain water. This water is usually neutralized by reactions with minerals in the soil zone, but has the potential to affect transport of many species.
Acidic water acts to mobilize chemical constituents in a soil. Common chemical reactions involve carbonate and sulfate minerals. Dissolution or precipitation of carbonates changes the water pH and to some extent the TDS content of the soil water, and dissolution of sulfates causes an increase in the TDS. In addition, low pH water is more likely to mobilize metals or other compounds such as ammonium that might be attached to clays or colloids.
Irrigation water is a potential source of recharge. This water may come from surface water, such as rivers, or ground water, the more common source in Kansas. The irrigation water is pumped from the source to the area to be sprayed or flood-irrigated. Depending upon the region's climate, evapotranspiration may change the chemistry of the water prior to its movement into the vadose zone. Evapotranspiration concentrates the dissolved species in water because of simple evaporation and/ or because of water use by plants (transpiration). Whereas evaporation concentrates all dissolved species in water, transpiration is selective, concentrating only those constituents not used by the plants. In either case, many chemical constituents in the soil water become more concentrated than in the source water used for irrigation. This concentrated water migrates through the vadose zone and interacts with the soil, thereby recharging an aquifer with water containing more dissolved solids than expected. The end result can be a degradation of water quality over time (Deverel and Fujii, 1990; Parker and Suarez, 1990; Tanji, 1990).
Evaporation of the water during irrigation of cropland may increase the TDS content in the underlying ground water over time. In addition, use of agrichemicals can contaminate ground water by transporting chemicals from the surface to the ground water. Irrigation thus influences assessment of safe yield from the water-quality perspective, as well as having a direct influence on withdrawal of water from an aquifer. The following sections describe two examples in Kansas where irrigation has probably caused deterioration of water quality in an aquifer.
South Fork, Beaver Creek--In Sherman County, Kansas, the Ogallala aquifer in the vicinity of the South Fork of Beaver Creek (fig. 5.1) is contaminated by nitrate, with concentrations above the U.S. Environmental Protection Agency drinking-water limit of 10 mg N/L (fig. 5.16; Townsend, 1995). This area was farmed in sugar beets from the 1950's to early 1980's. Sugar beets are a high fertilizer-use crop (276-360 kg N/ha; 200-300 lb N/acre). Flood irrigation was used during this time, and the South Fork of Beaver Creek was used as the tailwater runoff route for irrigation water. The market for sugar beets collapsed in the early 1980's, and at that time much of the flood irrigation was replaced by more efficient center-pivot irrigation and different crops were grown.
Figure 5.16--Location map of wells sampled from 1989 to 1994 in Sherman County, Kansas. Graduated dots indicate level of nitrate-N concentration measured at each site (from Townsend, 1995).
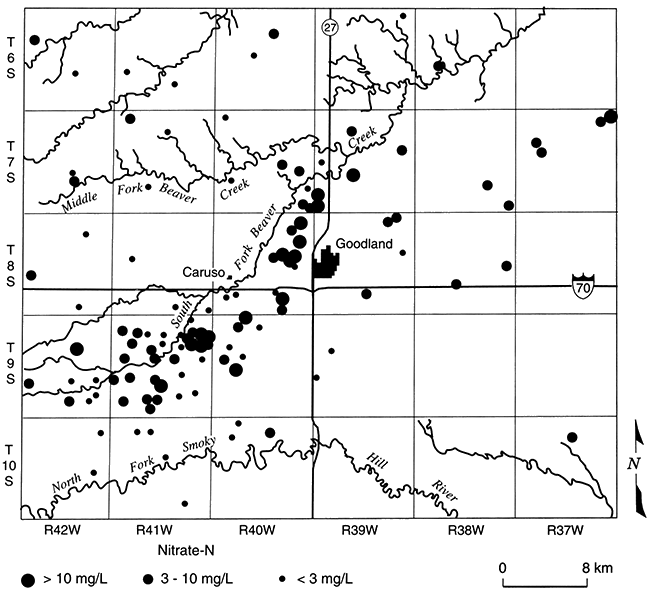
Ogallala aquifer water in this area now has a significantly higher total dissolved solids content than ground water in other parts of Sherman County. A working hypothesis is that the higher dissolved solids content is due to concentration of the salts in flood-irrigation water because of evaporation and transpiration. Comparison of water samples collected in 1978 with samples collected during a period from 1987 to 1994 shows that both total dissolved solids and nitrate-N have increased (fig. 5.17). This increase, well after the cessation of activity that may have created the problem, suggests a slow transport of water with higher total dissolved solids and nitrate from pre-1980's farming practices, and that water is now arriving at the irrigation wells.
Figure 5.17--Relationship of increased nitrate and TDS concentration is shown. Samples collected in 1978 have lower values than in more recently collected samples, suggesting continued concentration processes over time. Points shown on this plot are from the same well for the two collection periods or from wells in close proximity to each other for the two collection periods.
The mechanism for introduction of the concentrated irrigation water into the Ogallala aquifer in this part of Sherman County may not be simple transport through the unsaturated zone. The South Fork of Beaver Creek received the excess (tailwater) irrigation water from the sugar-beet fields, so that until the early 1980's it received high total-dissolved solids, nitrate-laden water some of the time. Farmers in the area recall the stream running bankfull during the irrigation season; water in the stream must have been composed almost entirely of the irrigation-return water. The streambed of the South Fork of Beaver Creek is approximately 30 m (l00 ft) above the groundwater table. Depth to water in nearby irrigation wells is considerably deeper (60 m [200 ft] or more) despite the only modest increase in ground elevation away from the creek (fig. 5.18). The increased head from the water in the stream and the gradient between the stream and the wells suggests that the South Fork of Beaver Creek is a losing stream for at least some times of the year, causing groundwater recharge through the stream bed. In this way, higher dissolved solids, nitrate-laden water from the irrigation tail water could have entered the Ogallala aquifer and travelled toward the irrigation wells in the vicinity of the creek.
Figure 5.18--Proposed recharge mechanism for nitrate-rich tailwater runoff to South Fork of Beaver Creek to reach the deeper Ogallala aquifer (Townsend, 1995).
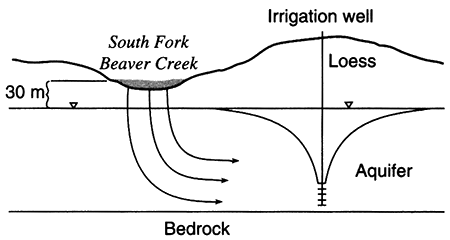
Arkansas River Basin-The Arkansas River corridor from the Colorado state line to Great Bend, Kansas (fig. 5.1), shows evidence of the effect of evaporation on ground-water quality. In this area, the concentration of total dissolved solids has increased significantly because of evapotranspiration of irrigation return flows to the river in both Colorado and Kansas (Whittemore, 1995b). Figures 5.19-5.21 show the elevated sulfate, nitrate, and chloride concentrations that occur in this area. In all three graphs the dashed line represents no change in concentration through time. The data points in each figure represent ground waters sampled in 1994 by the Department of Agriculture, which are the same irrigation wells or wells very close to irrigation wells sampled in 1975 by the Kansas Geological Survey.
Figure 5.19--Changes in sulfate concentration in waters from irrigation wells sampled in 1975 and 1994 in the Arkansas River valley (Whittemore, 1995b). Dashed line represents the condition in which concentrations were the same for both 1994 and 1975. Points above the dashed line indicate an increase in sulfate during the period; points below the line a decrease for the same period. Solid line represents U.S. EPA drinking-water limit of 250 mg/L.
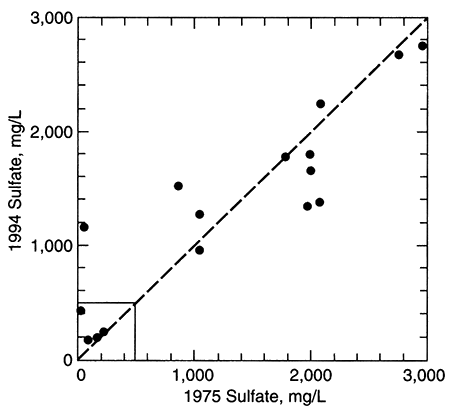
Figure 5.20--Changes in chloride concentration in waters from irrigation wells sampled in 1975 and 1994 in the Arkansas River valley (Whittemore, 1995b). Dashed line represents the condition in which concentrations were the same for both 1994 and 1975. Points above the dashed line indicate an increase in chloride during the period; points below the line a decrease for the same period. Solid line represents U.S. EPA drinking-water limit of 250 mg/L.
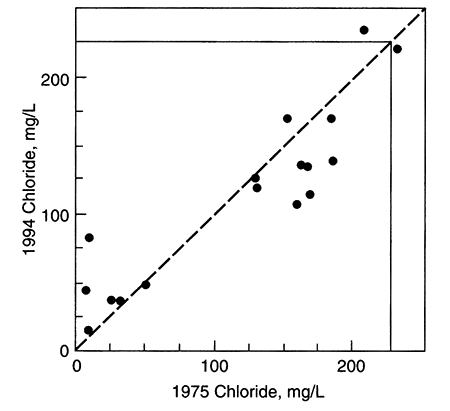
Increases in sulfate and chloride concentration from 1975 to 1995 (points above the dashed line) may reflect mixing of the relatively high-salinity Arkansas River water with ground water (fig. 5.19, 5.20). The data-point distribution for chloride and sulfate suggests two sources of chloride and sulfate (see The Consequences of Mixing, below). The spread of points supports the hypothesis of mixing between the river and the ground water, and suggests that the high-salinity river water is recharging the ground water. Increased ground-water-flow gradients caused by pumping of the many irrigation wells in the area is a likely reason for the river water moving into the alluvial aquifer adjacent to the river.
Decreases in sulfate and chloride concentration (points below the dashed line) generally occur in an area of southwestern Kansas that has a substantial percentage of land in the Conservation Reserve Program. Decreases may be related to land- and water-use changes (decreased irrigation perhaps resulting in decreased induced stream-aquifer interaction).
The nitrate-N graph (fig. 5.21) shows that many wells in the area have produced water with higher nitrate concentration in 1994 than in 1975. Because the area has been farmed for many years, it is unlikely that the source of nitrate is related to nitrification of organic nitrogen. The area is heavily irrigated, and the use of fertilizer with irrigation on an alluvial soil is likely to result in increased nitrate concentration in the ground water.
Figure 5.21--Changes in nitrate-nitrogen concentration in waters from irrigation wells sampled in 1975 and 1994 in the Arkansas River valley (Whittemore, 1995b). Dashed line represents the condition in which concentrations were the same for both 1994 and 1975. Points above the dashed line indicate an increase in nitrate during the period; points below the line a decrease for the same period. Solid line represents U.S. EPA drinking-water limit of 10 mg/L.
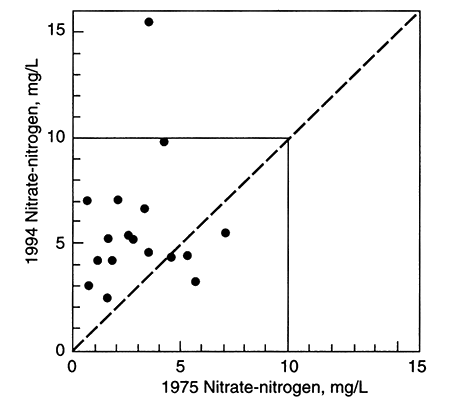
The overall decline in water quality in this area has occurred because of the shallow water table, irrigation return flows concentrated by evapotranspiration, and the low precipitation rate (35-40 cm/yr [14-16 inches/yr]) and recharge rate that might otherwise dilute the ground water.
Recharge can be affected by other fluids in a variety of situations. Point sources of contamination result from vertical flow originating from a specific, restricted location of contamination on the ground surface. Examples of point sources of contamination include landfills, storage tanks, ponds or lagoons, and areas at both commercial sites and farmsteads where chemicals are prepared by dissolving or diluting more concentrated stock. Contamination can also occur from more diffuse sources. This type, called nonpoint-source contamination, is one in which the source of contamination is so widespread that no single location can be identified as a sole source for the observed contamination. Examples of nonpoint-source contamination include widespread use of chemicals on cropland, road salt that dissolves in snowmelt and recharges shallow ground water, or nitrate contamination from lawn and golf-course fertilization.
Many different inorganic and organic chemicals are supplied from point and nonpoint sources. The two categories discussed below, landfills and agricultural chemicals, are representative of point and non point sources of contamination that may enter an aquifer. Planning for safe and sustainable production of an aquifer must take into account the possibility of such contamination affecting water quality of the ground water.
Landfills--Municipal landfills may be a source of inorganic contaminants such as nitrate, chloride, sulfate, iron, and manganese, and also a source of organic contaminants such as acetic acid and various volatile organic carbons (VOC's). These constituents are concentrated in the landfill environment and exit the landfill as leachate. Some components of the leachate react and some do not react with the surrounding aquifer, both cases resulting in degradation of ground-water quality. If a large volume of the aquifer is affected and/or pumped wells are nearby, then safe-yield determination of that aquifer is affected as calculations of capture zones of the pumped wells are made. A complication to interpreting the landfill-leachate effect on ground water is that the composition of some components of the leachate change with time (fig. 5.22). For example, many organic compounds are susceptible to degradation by bacteria, but the rate of degradation varies widely, making predictive models of the effects of landfills on ground water difficult. In addition, transport of metals such as iron and manganese depends upon the redox state of the water, so that transport of some metals is restricted to the area in which the leach-ate has created strongly reducing conditions (fig. 5.11).
Figure 5.22--Expected changes in selected components of landfill leachate with time (from Heck et al., 1992).
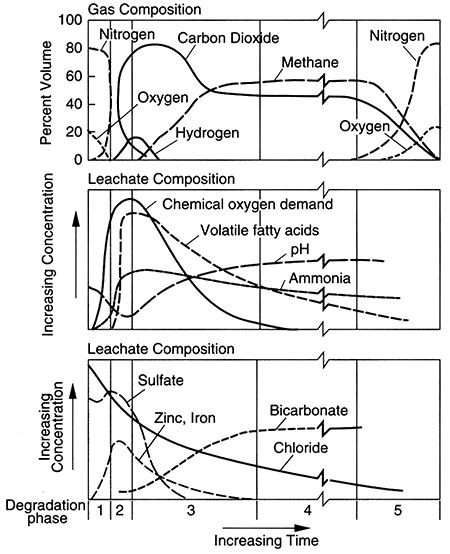
The following examples of contamination from landfills in Kansas represent different geologic and hydrologic settings common in Kansas. Ground-water contamination from landfills either is not common or at least is not well documented in Kansas at this time. The following examples show that although leachate contamination of ground water in Kansas does occur, positioning of landfills downgradient from water-supply wells has prevented serious contamination problems.
A study of the Reno County, Kansas, landfill (fig. 5.1; Heck et al., 1992) showed that dissolved organic compounds, some carcinogenic, have moved out of the landfill. This landfill is located on relatively impermeable silt and clay, and the contamination has been found in the underlying and more permeable sand and gravel aquifer. The landfill, however, is strategically located in that the contaminated water is moving toward Salt Creek, under which the ground water is naturally too salty to drink. For this reason it is unlikely that humans will inadvertently consume the ground water affected by landfill leachate. Furthermore, the Arkansas River lies between Salt Creek and the the city of Hutchinson. The sand-and-gravel aquifer affected by the leachate most likely discharges to the river. However, the distance to the river is enough that natural degradation of organic compounds and sorption of toxic metals is likely before discharge to the river.
The Sumner County, Kansas, landfill (fig. 5.1) is representative of the old and now illegal use of sand-and-gravel pits for waste disposal (Myers et al., 1993). The pit used for the Sumner County landfill was originally almost as deep as the water table, with the result that the landfill material placed in the pit is very close to the water table of the underlying aquifer. Investigation of the water quality in the area shows measurable concentrations of some organic and inorganic chemicals downgradient from the landfill, but the concentrations are probably relatively low because of mixing with the relatively fast-moving, clean ground water in the sand-and-gravel aquifer.
Landfills placed on rock that has fracture porosity also can affect ground-water quality because of the relatively rapid transport through fractures. The rapid transport reduces the amount of natural degradation and sorption that occurs before leachate enters ground water. The City of Olathe, Kansas, landfill, in Johnson County, eastern Kansas (fig. 5.1; Rasmussen et al., 1994) was excavated to fractured limestone and shale bedrock before being filled. At this site, much of the produced leachate found down gradient of the landfill will likely discharge to nearby creeks because of the geology and topography at the site. Flow paths are difficult to determine in such a setting, and degradation of surface-water quality is more of an issue than ground-water quality.
From these cases, it is clear that landfills have the potential to affect ground-water quality. Landfill design, age, geologic and topographic setting, and proximity to usable ground water all determine the impact of the inevitable escape of leachate from a landfill. Landfills may represent a threat to ground-water quality, and thus must be considered in determination of safe yield of an aquifer.
Agrichemicals-Atrazine--Fertilizers, pesticides, and herbicides are agricultural chemicals that can lower ground-water quality. In many Midwestern states, herbicides are more commonly used than pesticides, and atrazine is the dominant chemical used in areas growing corn, soybeans, and grain sorghum (Burkart and Kolpin, 1993). The escape of herbicides from cultivated land is apparently even more regional: in the western part of the Midwest (fig. 5.23), more herbicides, particularly atrazine and its metabolites (degradation products), are found in the surface and ground water than in the eastern part. In Kansas much of the work evaluating the occurrence of herbicides has focused on surface water, particularly in eastern Kansas where corn, sorghum, and soybeans are grown (e.g., Pope et al., 1996). The annual precipitation rate and patterns, topography, geology, and soils in this area create a region in which more precipitation ends up as overland flow than infiltration to the ground water. As a result, atrazine concentrations are generally higher but seasonally dependent in the streams and more constant in lakes and reservoirs (Pope, 1995; Stamer et al., 1995). Well production that results in stream-to-aquifer flow paths could therefore cause movement of atrazine-contaminated surface water into an adjacent aquifer.
Figure 5.23--Sampling sites (all triangles) in the Midwest during a survey for herbicides in surface and ground water. Filled symbols show locations of samples with detectable herbicide, mostly atrazine and its metabolites (from Burkart and Kolpin, 1993). Note the higher number of positive detections in the western part of the region.
Investigations of atrazine concentrations in ground water in Kansas are so recent that most studies are surveys of wells in order to assess the extent of the problem. One of the earliest surveys, by Steichen et al. (1988), found that four wells sampled in a random sampling design across the state (l00 wells total) had atrazine concentrations above the 3-ppb drinking-water limit. In addition, eight wells in the study had pesticide(s) detected in the well water. The herbicide and pesticide results contrast with indicators of fertilizer-affected ground water: 28 wells had nitrate-N above the drinking-water limit of 10 mg/L.
A recent statewide study of atrazine concentration in various aquifers in Kansas showed that approximately 72% of 84 wells sampled (including repeated sampling of the same wells) had atrazine concentrations below the detection limit of 0.10 ppb, 24% of the wells had concentrations between 0.10 and 1.5 ppb, and 4% of the wells were greater than the 3-ppb EPA drinking-water limit (Townsend et al., 1997). Of the wells with atrazine above the drinking-water limit, all were found to be affected by point-source contamination.
In another survey done by Perry and Anderson (1991), 111 irrigation wells were sampled. Only 5% of these wells had detectable atrazine, and all concentrations were below the drinking-water limit. The areas with detectable atrazine in ground water have shallow water tables and coarse-textured soils that may have facilitated the movement of atrazine to the ground water. Of the wells sampled, nine had nitrate-N above the drinking-water limit. The nitrate, from agricultural fertilizer, proves that water affected by agricultural practices is reaching the ground water. Therefore, the small number of wells affected by atrazine suggests that all agrichemicals cannot be expected to behave the same way during transport from the land surface to the water table.
The persistence of atrazine is apparently affected by a number of factors, some or all of which may affect the persistence of other agrichemicals, such as nitrate, differently. Application rates determine the loading of herbicide to the land surface, irrigation influences the amount of movement below the root zone, vadose-zone stratigraphy determines rate of transport through the unsaturated zone, and bacterial degradation of the atrazine determines whether or not atrazine "survives" during water transport through the unsaturated zone (Sophocleous et al., 1990). The ratio of one degradation-product concentration to the atrazine concentration may be a useful indicator of residence time in the soil zone. Short residence time indicates efficient "bypasses" for water through the soil zone (such as macropores) or proximity to source, such as in point-source instead of nonpoint-source contamination (Thurman et al., 1992; Squillace et al., 1993).
Investigation of the fate of herbicides in the unsaturated zone and in ground water, therefore, is really just beginning. Safe and sustainable pumping rates for aquifers must take into account arrival time of recharge that may be tainted with herbicides or other agrichemicals. Guidelines for areas susceptible to such contamination should certainly include consideration of agricultural practices, soil type (including description of transport paths, especially macropores), and climate and irrigation practices, as well as more specific information about degradation products of agrichemicals and their production rates.
Recharge may occur from an underlying aquifer (possibly containing more saline water) to an overlying aquifer (usually containing fresher water). A hydraulic gradient between units favors upward flow when, for example, natural pressure gradients are higher in a deeper aquifer or when drops in the water table or potentiometric surface in the shallower aquifer are induced by extensive pumping. Several areas in Kansas demonstrate interaquifer flow that results in poor-quality water entering a freshwater aquifer.
Effects of Natural Gradients on Upward Recharge--Natural hydraulic gradients cause movement of poor-quality water from a deeper aquifer (Permian) into an overlying freshwater Dakota aquifer system (fig. 5.1). Areas in the Dakota where this occurs correspond to areas where intervening confining units have pinched out (see Chapter 4 of this volume). The distribution of dissolved chloride in Dakota aquifer water shows that very high chloride is found in a beltlike region trending approximately northeast-southwest and paralleling the Dakota outcrop. The western part of this region coincides with areas where the Dakota is in hydraulic connection with a deeper aquifer containing saline water, the Cedar Hills aquifer (fig. 5.24; Macfarlane et al., 1989). The higher hydraulic head in the deeper aquifer causes saline-water discharge into the Dakota aquifer, rendering the water unpotable. The eastern part of the area containing saline water demonstrates that a considerable part of the Dakota aquifer downgradient from the zone of leakage has been affected by the saline fluid discharging into the Dakota along a fairly narrow corridor.
Figure 5.24--Dakota aquifer, Kansas. A) Patterned area shows where the Dakota and underlying Cedar Hills aquifers are hydraulically connected (modified from Macfarlane et al., 1989). B) Contours of chloride (mg/L) in the Dakota aquifer, Kansas. Heavy line is the trace of the outcrop or subcrop of the Dakota aquifer. Where the Dakota is hydraulically connected to the Cedar Hills, higher chloride concentrations are present.
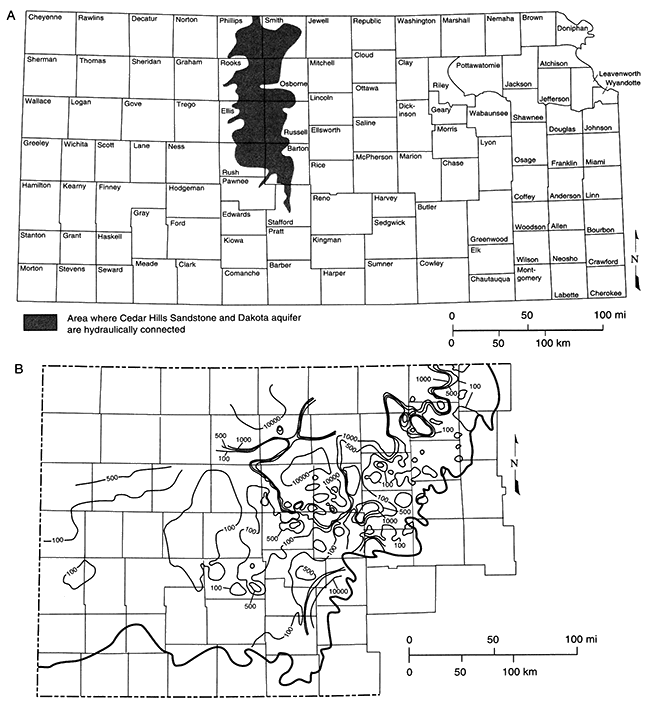
Effects of Pumping on Upward Recharge--In south-central Kansas, numerous areas of saltwater intrude from the underlying Permian aquifer into an overlying unconsolidated freshwater alluvial aquifer, especially in the Great Bend Prairie region (fig. 5.1). Current work in salt-affected areas indicates that the fluctuation of water chemistry (chloride, nitrate-N, and total dissolved solids) may be strongly affected by pumping patterns in the alluvial aquifer above unconfined Permian bedrock. When pumping begins, nitrate-N concentrations (the source for which is presumed to be fertilizer from the land surface) are high but decrease with increased pumping time. Chloride concentrations (the source for which is the saline Permian aquifer) are low at the beginning of pumping and increase during the irrigation season (fig. 5.25; Young, 1995). Nonsteady changes in concentration of these components occur with time, suggesting that there is mixing of stratified sources of water, the degree of which depends upon the size of the cone of depression that develops around the pumping well.
Figure 5.25--Chloride and nitrate-N concentrations for wells at Siefkes site in Stafford County, Kansas. Note the effect of pumping on increasing chloride concentration over time and decreasing nitrate-N concentration over time (after Young, 1995).
In addition to fluctuations in water chemistry, fluctuations in the water levels of the freshwater aquifer and the Permian aquifer are observed throughout the irrigation season (fig. 5.26; Young, 1995). At times during the irrigation season, the water level in the deeper portions of the freshwater aquifer falls below the water level of the Permian aquifer, indicating that the underlying aquifer is under higher pressure than the overlying alluvial aquifer. There is most likely slow, upward, natural discharge from the Permian aquifer at all times, but upward discharge rates are probably increased because of pumping of irrigation water from the shallow aquifer and the consequent drop in hydraulic head.
Figure 5.26--Hydrographs of Permian (P), deep-aquifer (DA), and water-table (IM) monitoring wells at Siefkes site. Note that Permian water levels rise above water-table (IM) levels after extended periods of pumping (after Young, 1995).
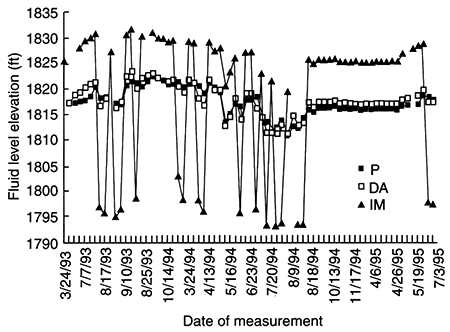
In some areas, the total dissolved nitrogen in Permian aquifer water is higher than in the overlying alluvial aquifers. Dissolved nitrogen in the Permian aquifer ground water is in the reduced form, ammonium (see Boxed section 5.3, The Nitrogen Cycle), and does not result from agricultural-fertilizer contamination. Upward movement of saline Permian-aquifer fluid, especially as accelerated by pumping of the overlying aquifer, results in addition of nitrogen species to the shallower alluvial aquifer. The nitrogen species are then mostly oxidized to nitrate in the more oxidizing environment of the alluvial aquifer (Whittemore, 1993). Therefore, distinctions must be made between two situations causing increased salinity accompanied by elevated nitrate concentrations, the first resulting from evapotranspiration and the second from dissolution of Permian salt accompanied by naturally occurring elevated nitrate concentrations. These distinctions are important to planning for sustainable production rates of an aquifer in order to minimize the intrusion of saline fluids into a freshwater aquifer.
The pumping of wells causes a reduction in pressure within an aquifer that reverses normal ground-water flow. Because gradations in the chemistry of ground water typically occur within an aquifer, mixing of chemically different water can cause chemical reactions in the mixed fluids (see Chemical Reactions in Ground Water). Furthermore, the movement of fluid implies that the fluid must readjust to a different part of the aquifer. The moving fluid may have been in equilibrium with a different type of rock, under different redox conditions and temperatures, or may have been naturally of a different chemistry because of other factors. This fluid reacts with the new environment by dissolving or precipitating solid phases and being involved in sorption or desorption reactions. Besides reacting directly with the solid matrix, the introduced fluid also mixes with the resident fluid, the proportion of which can determine the extent of mineral reactions that occur.
Vertical stratification of fluids in aquifers has been observed in many aquifers, introducing the expectation that pumping a well should mix chemically different fluids. The amount of chemical stratification is a response to the heterogeneity of the aquifer (see Glossary, heterogeneity) as well as the hydrodynamics of recharge and throughflow. A theoretical, completely homogeneous aquifer with a large hydraulic conductivity is expected to have relatively homogeneous water chemistry in all climatic settings except arid because residence time of the water in the aquifer is relatively short. All other types of aquifers should be chemically stratified. One example of vertical chemical gradients is found in the alluvium of the Kansas River near Lawrence, Douglas County, Kansas (figs. 5.1 and 5.27; Macpherson et al., 1996), where the gradient is not smooth but contains peaks and valleys throughout the profile. Another example, from the Great Bend Prairie alluvial aquifer (fig. 5.1), shows an extreme case of vertical chemical stratification in which saline fluids from salt dissolution are "puddled" at the base of the alluvial aquifer (fig. 5.28; Garneau, 1995; Buddemeier et al., 1992).
Figure 5.27--Variations in chloride (left) and magnesium (right) with depth in Kansas River alluvium near Lawrence, Douglas County, Kansas. The profiles demonstrate chemical stratification in an alluvial aquifer, probably controlled by permeability differences in the sediments.
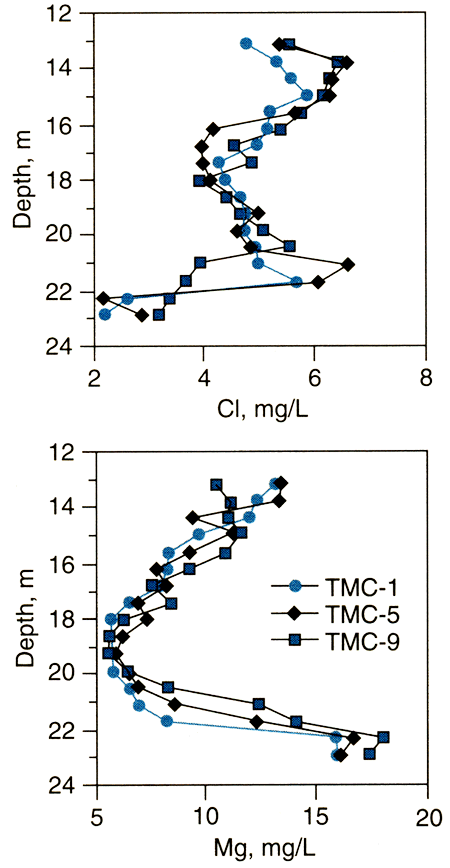
Figure 5.28--Clay lenses (gray) and saltwater (medium blue) in the Great Bend Prairie alluvial aquifer, Stafford County. This west-east cross-section shows estimated locations of discontinuous clay lenses (from gamma ray and focused induction well logs) and shows upconing of saltwater (especially site 17) from bedrock (blue-gray) attributed to pumping of a well. Deeper water, in this case saline, is drawn upward into pumping wells (modified from Garneau, 1995).
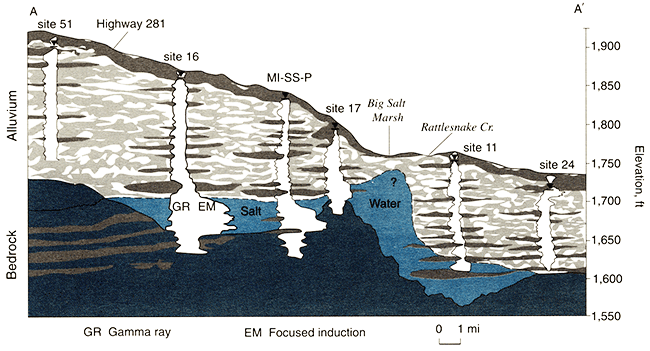
Patterns and extent of lateral variations in water chemistry within an aquifer are much better documented than vertical variations, being first linked to specific chemical processes by Foster (1950). Progressive change during recharge of dilute rainwater into an aquifer and subsequent movement along ground-water-flow paths is caused by mineral dissolution/precipitation, ion exchange, sorption, and adjustments due to changes in redox state in the aquifer. It is important to note that ground-water movement is typically very slow. In addition, the pore space that contains the water in an aquifer normally accounts for up to a maximum of 30% of the total volume in a lithified aquifer and perhaps 40% in an unconsolidated one. For these reasons, the chemistry of ground water is usually determined by the composition of the aquifer matrix, i.e. the minerals in the rock that hosts the aquifer. Furthermore, differential ground-water velocities in a typical heterogeneous aquifer result in water in the pores of the coarser-grained parts of the matrix having a chemistry different from water in the pores of the finer-grained parts. This spatial variation in water chemistry becomes more complex as the amount of aquifer heterogeneity increases.
Both vertical and lateral water-chemistry variability provide the opportunity for waters of different chemistry to mix when an aquifer is pumped. The lowering of hydraulic head that causes water to be produced from a well creates hydraulic gradients in the vicinity of the well that affect the aquifer according to the hydraulic conductivity of the rock or sediment. In the normal case of aquifer heterogeneity, although most of the water produced from a well comes from the zones of highest hydraulic conductivity, some will be drawn from the lower-conductivity zones, causing mixing of fluids from the different zones. Identifying mixing of different fluids can be straightforward if no chemical reactions result from the mixing, or more complicated if reactions remove or add dissolved species and mask the compositions of the pure end members. Although some scientists use statistical techniques to identify mixing (Christophersen and Hooper, 1992), there are simple cases in which mixing is relatively easy to identify.
When chemical reactions do not occur as the result of mixing, the end-member fluids often can be identified by plotting two components of the solution on bivariate plots and observing the relationships. Two end members with no mixing plot as separate clusters of points (fig. 5.29), two end members mixing with no chemical reaction plot as linear arrays (fig. 5.30), three end members mixing with or without chemical reaction are evident when a triangular region encompasses the spread of the data points (fig. 5.31), or the data fall on a linear trend but are clustered instead of continuous (fig. 5.32). Mixing can also be deduced from a plot of the concentrations of all of the determined ions in solution when plotted on a Schoeller diagram (fig. 5.33). This simulation demonstrates dilution of a saline water, evident by the parallel lines of the same pattern. A bivariate plot on which one variable is a ratio of two or more variables and the other is a single variable will show a curvilinear array of data points when two-member mixing occurs (fig. 5.34).
Figure 5.29--Eight springs plot in two distinct clusters, suggesting coexistence of two chemically different waters that have not mixed. Concentrations are in meq/L (from Mazor, 1991; figs. 5.29-5.34 reprinted by permission of John Wiley & Sons, Inc.).
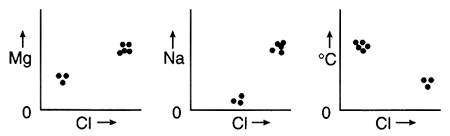
Figure 5.30--Spring water (different from fig. 5.29) shows mixing without chemical reactions, suggesting a saline, hot end member and a fresh, cool end member. Concentrations are in meq/L (from Mazor, 1991).
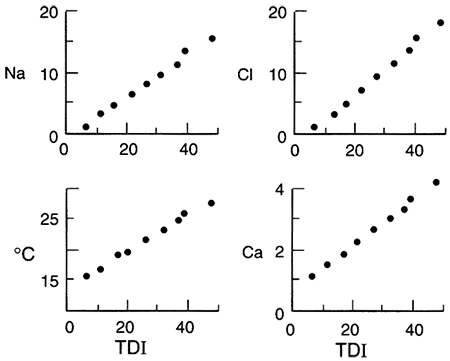
Figure 5.31--Triangular areas on compositional diagrams form where three distinct water types intermix in varying proportions (from Mazor, 1991).
Figure 5.32--Where three distinct waters exist in a region but do not mix, isolated clusters of data appear on scatter plots such as these (from Mazor, 1991).
Figure 5.33--Dilution of a saline water (#5) with a dilute water (#1) is easily visualized on a Scoeller diagram such as this (from Mazor, 1991).
Figure 5.34--A plot of a single variable, such as chloride, versus a ratio variable, such as the oxygen isotope ratio Δ18O (units of per mil, ‰), suggests mixing of waters when data from the same region or even the same aquifer fall along a curve. In this case, coastal wells near the Gulf of California show an effect from sea-water intrusion (from Mazor, 1991).
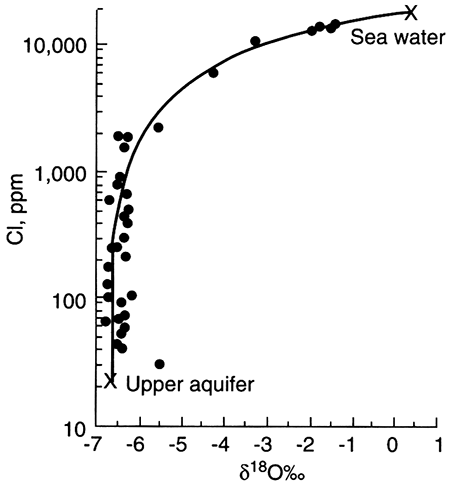
Mixing of fluids frequently causes chemical reactions to occur. The reactions result from several chemical phenomena. First, nonlinear mineral-solubility relation results in a mixed fluid that is over- or undersaturated with respect to a mineral even when the fluid end members were saturated. Second, when a mixed fluid has a TDS higher or lower than the fluid end members, the mixture will become undersaturated (corrosive) or oversaturated (encrusting) with respect to mineral phases (e.g., Freeze and Cherry, 1979). This phenomenon results from ions being chemically less "reactive" (having a lower "activity") in a high-TDS fluid than in a low- TDS fluid. Third, the common-ion effect results in precipitation of a mineral when the fluids have an ion in common but at different concentrations because of equilibration with different minerals. For example, a fluid (fluid "A") in equilibrium with the soluble mineral gypsum contains high concentrations of calcium. Fluid "A" can also be in equilibrium with calcite, which also contains calcium, as long as the carbonate concentration of the fluid is very low (the product of the calcium content and the carbonate content, or [Ca+2]*[CO3-2], is a constant at any temperature). A fluid "B," in equilibrium with calcite but never having been in contact with gypsum, will contain only small amounts of calcium and equivalent amounts of carbonate. A mixture of fluids "A" and "B" will be oversaturated with respect to calcite, because the product, [Ca+2]*[CO3-2], will be larger than the equilibrium value as a result of the high calcium content of fluid "A." Finally, mixing may also cause sorption or desorption of trace elements or organics from surfaces, or changes in ion-exchange equilibrium as well as changes in redox potential.
Besides chemical reactions that cause a mixed-fluid chemistry to be different from the simple proportion of the end-member fluids, the unexpected coexistence of water "tags" can provide insight into the introduction of water to a new part of the aquifer. For example, the simultaneous presence of tritium (3H), indicating water recharged within the past 40 years or so, and very small amounts of carbon-14 (14C; see Glossary, isotope, and Boxed section 5.2, Isotopes and Ground Water), indicating very old water, suggests the addition of very dilute, recently recharged water to older water. Other indicators of mixing may not be present, but the presence of indicators of water recharged in the distant past in the same fluid with indicators of water recharged in the recent past is a fairly robust argument for the mixing of old water with new. Similarly, compounds added to the atmosphere from manufactured products, such as chlorofluorocarbons formerly used as a propellant for spray cans, also are useful indicators of recently recharged water (Dunkle et al., 1993).
Selected examples of mixing of fluids and the consequences of mixing are given below. These were chosen on the basis of their known or anticipated relevancy to Kansas aquifers, but the examples given do not cover all the possible types and consequences of mixing fluids, either in Kansas or elsewhere.
Example--Bromide/chloride ratio, a tracer of water sources--An example of the usefulness of a bivariate plot for identifying sources of water in Kansas is given by Whittemore (1995a). In this method, the ratios of a minor halogen element, bromide (Br-), to a major halogen element, chloride (Cl-), when plotted against chloride fall in distinct, restricted areas of the graph for different waters. These water end members attain a distinctive Br/Cl ratio and chloride concentration through inheritance (such as a sea-water signature or a rainwater signature) or water-rock interaction (such as dissolution or reprecipitation of halite, Holser, 1979; or breakdown of organic matter and consequent release of Br, Macpherson, 1994). Whittemore gives an example of freshwater in the Great Bend Prairie aquifer (fig. 5.1) mixing with saline water from the underlying Permian units, the signature of which is a fairly narrow, curved, mixing corridor between low chloride, high Br/Cl and high chloride low Br/Cl end members (fig. 5.35). The saline water results from dissolution of the mineral halite (rock salt) and is distinctive from oil-field brines produced in the area: the oilfield brines have higher Br/Cl ratios as well as high chloride content. Mixtures of the three end-member fluids in this area, the oil-field brines, freshwater, and saltwater from halite dissolution, plot along distinct curves or in limited envelopes. Analysis of samples that are intermediate to the end members allows a semi-quantitative assessment of the proportions of each of the end members in the mixture (fig. 5.35).
Figure 5.35--Br/Cl mass ratio-chloride concentrations are distinctive for different types of water in Kansas. Halite-dissolution water contains high Cl and low Br/Cl (P's on the plot). Freshwater (derived from rainwater) has low Cl and high Br/Cl (A's in the upper left on the plot). Oil-field brines shown here have high Cl and intermediate Br/Cl (O's on the plot). Mixing of freshwater and salt-derived water plots below the solid curved line; a mixture of the three water sources plots on any of a whole family of curves, some of which are shown here as dashed. The proportion of the three end members can be approximated from the position on the mixing lines (from Whittemore, 1995a).
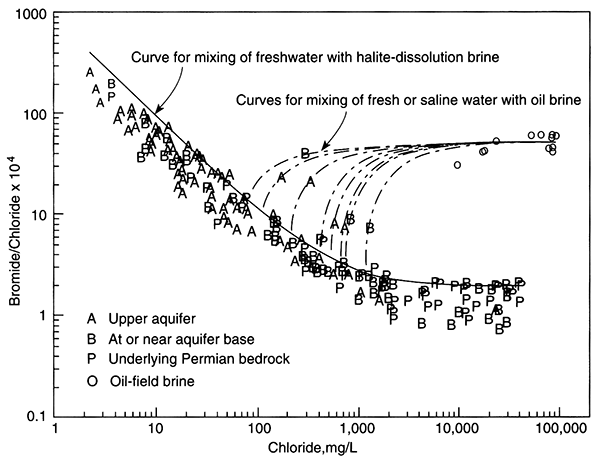
Example--Mixing and corrosion--One of the best known consequences of the mixing of different waters is that of two waters, both of which are saturated with respect to calcite but are at equilibrium with different partial pressures of carbon dioxide. The relationship between the amount of Ca+2 and partial pressure of CO2 in solution at equilibrium with calcite is not linear (fig. 5.36). Mixing of fluids labeled A and B on the plot results in a fluid composition found on the line connecting these two points. Because of the curvature of the equilibrium line, the mixed fluid, no matter the proportion of A and B, is undersaturated with respect to calcite and will dissolve calcite. This phenomenon is thought to be one of the reasons for the formation of karst features in limestones (e.g., Thrailkill, 1968) and may account for much of the widening of fractures in limestone aquifers that makes them productive aquifers. This effect is not exclusive to limestones: modeling of the Dakota aquifer in Kansas (which has carbonate cements) has suggested that mixing of saline fluids from the underlying Cedar Hills Sandstone aquifer with Dakota aquifer water causes undersaturation with respect to the carbonate minerals (fig. 5.37; Macfarlane et al., 1989). The implications of this phenomenon are very interesting: mixing of a small amount of saline water with a more dilute aquifer water through intrusion, either natural or induced, causes the mixed fluid to become corrosive with respect to the carbonate minerals. Because carbonates are very common, the corrosive fluid will dissolve part of the aquifer matrix, causing it to become more porous and permeable, allowing more saline water to intrude. In this way, the zone of intrusion can propagate and through time affect a large part of an aquifer.
Figure 5.36--If two waters (a and b) that are in equilibrium with calcite (fall on the heavy line) mix, the resulting mixture (anywhere on the light line) is undersaturated with respect to calcite and will dissolve calcite (from Drever, 1988; reprinted by permission of Prentice-Hall, Inc.).
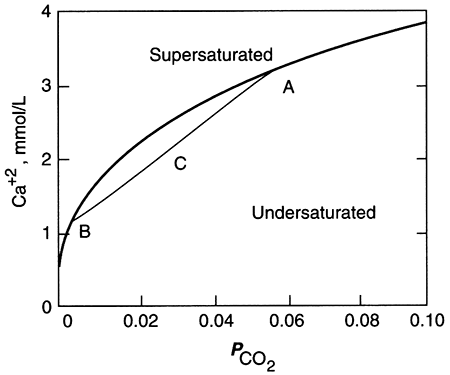
Figure 5.37--The saturation state of three carbonate minerals along one ground-water flow path in the Dakota aquifer, Kansas. A saturation index of zero (0) is perfect saturation; negative saturation indices indicate undersaturation and positive saturation indices indicate oversaturation. The Dakota aquifer ground water becomes undersaturated (saturation index <0) with respect to the carbonates downgradient of the Cedar Hills Sandstone subcrop. Leakage of water from the Cedar Hills Sandstone and the subsequent mixing causes the undersaturation (Macfarlane et al., 1989).
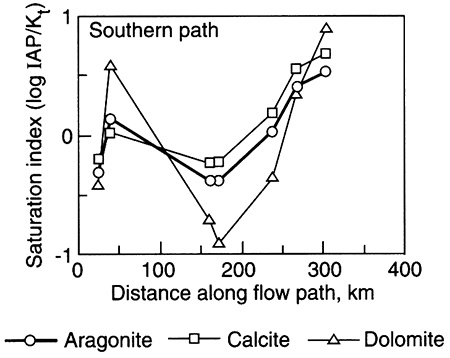
Mixed ground water may become corrosive or encrusting with respect to other minerals, as well. A theoretical example is given next, demonstrating the effect of the mixing of waters with different iron content and different Eh conditions (see Boxed section 5.1).
Example--Dissolved iron and mixing--Iron is a redox-sensitive metal that is fairly commonly found in ground water. Iron exists in two oxidation states in water, ferrous iron, FeII, having a valence of +2 and ferric iron, FeIII, having a valence of +3. FeIII is the dominant form of dissolved iron under the unusual conditions of very acid pH and very high pe. FeII dominates in more typical conditions of less acid to very alkaline pH and moderately high to very low pe (fig. 5.38). The common and mature oxide-mineral phases that contain iron are hematite (Fe2O3) with ferric iron and magnetite (Fe3O4) with both ferric and ferrous iron. These minerals, along with pyrite (FeS2), are often the primary sources of iron in solution. (In some aquifers, iron-rich alumino-silicate minerals such as pyroxenes and amphiboles also are a source of iron.) Iron typically precipitates from solution into solids that are not true minerals but types of amorphous oxyhydroxides such as Fe(OH)3 and Fe(OH)2 or FeOOH. Because these precipitates are very fine grained, total iron in a solution, without filtering, is often dominated by colloids of iron oxyhydroxides that are suspended in the solutions (Whittemore and Langmuir, 1975).
Figure 5.38--Stability fields of iron oxides, iron carbonates, and iron sulfides. Conditions chosen to create this diagram are: temperature of 25°C, 1 atmosphere total pressure, total dissolved sulfur of 10-6 mol/L (if all sulfur was sulfate, concentration would be about 0.1 mg/L), total dissolved carbonate of 1 mol/L, total dissolved iron of 10-6 (heavy lines) or 10-4 (light lines; equivalent to about 0.06 or 6 mg/L, respectively). (From Garrels and Christ, 1965.)
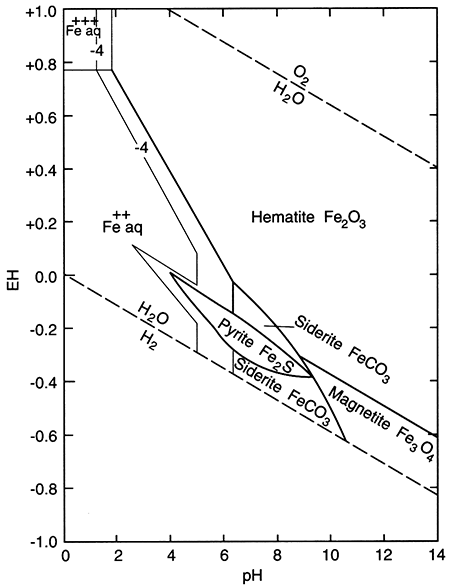
For ground water in a steady state, one can fairly safely assume little change in the water chemistry through time. Development of an aquifer can result in the mixing of waters from different aquifers or from different parts of an aquifer, with the result that chemical reactions are accelerated and water chemistry different from what is expected from simple mixing can result. Mixing of ground water from the Tonganoxie aquifer (Douglas Group) and the Kansas River alluvium in Douglas County could occur where Kansas River alluvium overlies the Pennsylvanian sandstone, in Douglas and Leavenworth counties (fig. 5.1). Computer mixing of published water analyses (from Miller et al., 1991) using the geochemical speciation program PHREEQE (Parkhurst et al., 1990) shows that a mixing of Kansas River alluvium ground water with Tonganoxie aquifer water causes a dramatic change in the saturation state with respect to the amorphous iron hydroxides (fig. 5.39). A mixture of about 10% Tonganoxie ground water with 90% alluvium ground water is the crossover point for strong oversaturation with respect to iron oxyhydroxides (the case for pure alluvium ground water) to strong undersaturation (the case for pure Tonganoxie ground water). The crossover point is not always at a 9: I proportion, but is determined by how well buffered each of the mixing end members are relative to each other. In this case, the Tonganoxie ground water is more strongly buffered against redox changes (that in turn control saturation with respect to iron-bearing minerals) than the alluvium ground water.
Figure 5.39--Theoretical mixing of Tonganoxie aquifer water and Kansas River alluvium ground water. In this case, mixing in the proportion of about 9 parts to 1 part results in a dramatic change in the mixture's tendency to dissolve (log SI < 0) or precipitate (log SI > O) ion compounds.
Recharge of an aquifer affects the quality as well as the quantity of ground water in an aquifer. Land-use practices and point- and nonpoint-source contamination may affect water quality of surface-derived recharge directly. Natural variations in soil permeability can change the quality of ground-water recharge through the unsaturated zone. Finally, enhancement of cross formational flow or flow from low-permeability zones within aquifers can affect water quality. In all cases, physical and chemical processes influence the final water chemistry.
Impedance and enhancement of flow are physical processes in the unsaturated zone that affect the chemistry of the water. Impedance of flow increases the residence time of water, allowing evapotranspiration and/or chemical reactions to change water chemistry. Enhancement of flow occurs through fractures or macropores, providing a way for water to bypass the unsaturated zone and therefore not be affected by normal chemical and biochemical reactions that strip water of potentially harmful components. Vadose-zone stratigraphy describes the heterogeneity of sediments that affects flow rates through the soil zone.
Two kinds of chemical reactions common in the unsaturated zone are ion exchange and precipitation. These can affect clay minerals and result in decreased permeability. The lowered permeability not only lowers the quantity of water that recharges the aquifer, but also increases the water residence time in the soil, permitting changes in the chemistry of the recharge water.
Recharge from a saline aquifer beneath or adjacent to a water-supply aquifer can result in deterioration of water quality if pumping practices result in a mixing of the two waters. In addition, the mixing of fresh and lesser quality waters can occur from movement upward because of artesian pressures in a lower aquifer or natural discharge points because thinning of an aquifer abuts an adjacent freshwater aquifer.
Degradation of water quality can also occur because of pumping when there is lateral variation of water chemistry. In fact, lateral changes are to be expected, as are various types of chemical stratification that are dependent upon heterogeneity in the aquifer hydraulic conductivity.
Some generalities can be made about expected changes in water quality because of mixing induced by pumping. The upconing of water from deeper parts of an aquifer or from cross formational flow from deeper aquifers will usually result in the introduction of saline fluids with a composition dominated by sodium (Na) and possibly calcium (Ca) and chloride (Cl), Introduction of rainwater concentrated by evaporation or evapotranspiration may also be saline and in some cases may be sulfate-rich. Irrigation water concentrated by evaporation or evapotranspiration may contain high levels of nutrients such as nitrate and other agrichemicals, as well as being more saline versions of the ground water or surface water used for irrigation. Movement of fluids from point sources such as landfills, storage tanks, and ponds into an aquifer can be identified by studying the fluids at the point source, but in general may be rich in dissolved organic carbon (DOC) and possibly metals.
Mixing of fluids may be simple, in that the mixed fluid composition is in simple proportion to the end members. More commonly, a mixed fluid becomes corrosive or encrusting with respect to selected minerals, the best documented being calcite and the iron minerals. Furthermore, a mixed fluid may undergo ion exchange with nearby clay minerals because the proportions of ions in the mixture are no longer in equilibrium with the clays. The resulting fluid may not necessarily be recognizable because of loss or gain of dissolved species as the fluid comes into equilibrium with its surroundings. The use of conservative and refractory species, those that tend to not react with the matrix, allows identification of end members in many cases. Examples of identifiers used successfully include Br/Cl ratios and isotopes.
The management of an aquifer or a system of aquifers must include consideration not only of yield (the volume of water) but also the quality of water to be produced. It is the rule, not the exception, that stratification of chemistry occurs within as well as between aquifers. Only very homogeneous aquifers that are well flushed (because they have high hydraulic conductivity and are found in climates that are temperate to humid) are likely to have homogeneous chemistry: these are exceedingly rare. The variation in water chemistry can occur on all scales. On a small scale, zones of lower and higher hydraulic conductivity contain slightly to very different chemistries of water over distances of centimeters. On a large scale, long-term flushing of an aquifer results in mostly lateral gradation in water quality over distances of 10's to IOO's of kilometers.
The entry of poor-quality fluids into an aquifer can occur through the unsaturated zone or from within or beneath the aquifer. Irrigation water can be concentrated through evaporation and add salinity, nutrients, or agrichemicals to ground water. Downgradient saline fluids within an aquifer can be drawn upgradient toward wells, or saline fluids at the base of an aquifer can be drawn upward to a well during pumping. Saline fluids from salt dissolution can leak upward into aquifers where confining beds are absent and hydraulic gradients favor such movement. This natural discharge can be accelerated through removal of fluids from the freshwater portion of the aquifer.
Water tends toward equilibrium with the solids with which it is in contact. Causing a fluid to move to a new position, therefore, may induce a chemical change. The change can occur because of mixing with another fluid, or because the fluid has come into contact with different mineral or organic phases. The changes occur because of several types of chemical reactions. A solid may dissolve and add dissolved components to the water or a solid may precipitate out of the water, removing solids. Ion exchange can occur when the ratio of cations in the fluid is not in equilibrium with the ratio on the clay minerals. Sorption or desorption can occur when the dissolved metals or organics are out of equilibrium with their respective components sorbed on the aquifer matrix. Species that are volatile may be removed from solution with a change in pressure or temperature, and species that are redox sensitive may change redox state, causing precipitation or dissolution of related mineral phases.
In Kansas, agriculture depends upon good-quality ground water and yet it indirectly or directly causes degradation of water quality through evaporation and the addition of agrichemicals. Recharge of this water through the unsaturated zone can eventually degrade the very source of water used for irrigation. The impact may be lessened somewhat by natural degradation processes in the unsaturated zone (for agrichemicals), but will not be totally eliminated.
In the Great Bend Prairie region and other regions of central Kansas, natural discharge of saline water from dissolution of buried Permian salt causes salinization of shallow alluvial aquifers. Population growth in these regions will result in the development of aquifers as a water supply. The movement of saline water into an alluvial aquifer is accelerated by pumping. In theory, the impact of saline-water movement could be managed so as to cause only a small portion of saline fluid to be mixed with the fresher water.
In the Dakota aquifer of Kansas, regional changes in water quality have been identified from the Colorado state line to the discharge areas in central Kansas. Ion exchange of calcium and magnesium for sodium causes a gradation in the ratios of these ions down the regional hydraulic gradient, as long-term flushing of the aquifer continues. The absence of a hydraulic barrier causes discharge of deeper, saline fluids from the Cedar Hills aquifer into the Dakota in selected regions, and a prominent zone of higher salinity fluids identifies those areas. Furthermore, in the Dakota in northwestern Kansas, saline fluids may represent the downgradient portion of an ancient flow system. These fluids have the potential to move into the freshwater portions of the Dakota if there is uncontrolled development of the Dakota as a ground-water supply. For each of these situations, aquifer development may induce movement of fluids into different parts of the aquifer and/or mixing, allowing chemical reactions to proceed as the fluids approach equilibrium with the solids.
Aquifer management should include assessment of the chemistry of fluids likely to move during the pumping of wells. The effects of mixing of the fluids and equilibration of the moving fluids with the aquifer can be predicted using simple to complex computer programs, depending upon the complexity of the problem and the amount of information available about aquifer parameters and chemical parameters. The outcome will necessarily be poor- to well-constrained predictions of the effects of aquifer development. Computer modeling of hydraulic-head changes in combination with modeling of the chemical changes that might occur is essential to the most efficient management of our water resources.
Appelo, C. A. J., 1994, Cation and proton exchange, pH variations, and carbonate reactions in a freshening aquifer: Water Resources Research, v. 30, no. 10, p. 2,793-2,806
Baedecker, M. J., and Back, W., 1979, Modem marine sediments as a natural analog to the chemically stressed environment of a landfill: Journal of Hydrology, v. 43, p. 393-414
Bass Becking, L. G. M., Kaplan, I. R., and Moore, D., 1960, Limits of the natural environment in terms of pH and oxidation-reduction potentials: Journal of Geology, v. 68, p. 243-284
Berner, E. K., and Berner, R. A., 1996, Global environment--Water, air, and geochemical cycles: Upper Saddle River, New Jersey, Prentice Hall, Inc., 376 p.
Berner, R. A., 1971, Principles of chemical sedimentology: New York, McGraw-Hili, 240 p.
Berner, R. A., 1975, Diagenetic models of dissolved species in the interstitial waters of compacting sediments: American Journal of Science, v. 275, p. 88-96
Bowen, R., 1988, Isotopes in the earth sciences: New York, Elsevier Applied Science Publishers, 647 p.
Brownlow, A. H., 1996, Geochemistry, 2nd edition: Prentice-Hall, Inc., Upper Saddle River, New Jersey, 580 p.
Buddemeier, R. W., Sophocleous, M. A., and Whittemore, D.O., 1992, Mineral intrusion--Investigation of salt contamination of ground water in the eastern Great Bend Prairie aquifer: Kansas Geological Survey, Open-file Report 92-25, 45 p.
Buol, S. W., Hole, F. D., and McCracken, R. J., 1989, Soil genesis and classification: Iowa State University Press, Ames, 446 p.
Burkart, M. R. and Kolpin, D. W., 1993, Hydrologic and land-use factors associated with herbicides and nitrate in near-surface aquifers: Journal of Environmental Quality, v. 22, no. 4, p. 646-656
Christophersen, N., and Hooper, R. P., 1992, Multivariate analysis of stream water chemical data--The use of principal components analysis for the end-member mixing problem: Water Resources Research, v. 28, no. 1, p. 99-107
Chu, Tyan-ming, 1995, Interpretation of geochemical evolution in the Dakota aquifer in Kansas based on coupled hydrogeochemical models: Ph.D. dissertation, The University of Kansas, Lawrence, 295 p.
Deverel, S. J., and Fujii, R., 1990, Chemistry of trace elements in soils and ground water; in, Agricultural Salinity Assessment and Management, K. K. Tanji, ed.: American Society of Civil Engineers, New York, p. 64-90
Domenico, P. A., and Robbins, G. A., 1985, The displacement of connate water from aquifers: Geological Society of America, Bulletin, v. 96, p. 328-335
Domenico, P. A., and Schwartz, F. W., 1990, Physical and chemical hydrogeology: New York, John Wiley and Sons, 824 p.
Dow, W. G., 1978, Petroleum source beds on continental slopes and rises: American Association of Petroleum Geologists, Bulletin, v. 62, no. 9, p. 1,584-1,606
Drever, J. I., 1988, The geochemistry of natural waters, 2nd edition: Prentice-Hall, Inc., Englewood Cliffs, New Jersey, 437 p.
Dunkle, S. A., Plummer, L. N., Busenberg, E., Phillips, P. J., Denver, J. M., Hamilton, P. A., Michel, R. L., and Coplen, T. B., 1993, Chlorofluorocarbons (CCl3F and CCl2F2) as dating tools and hydrologic tracers in shallow groundwater of the Delmarva Peninsula, Atlantic Coastal Plain, United States: Water Resources Research. v. 29, no. 12, p. 3,837-3,860
Fabryka-Martin, J., Whittemore, D.O., Davis, S. N., Kubik, P. W., and Sharma, P., 1991, Geochemistry of halogens in the Milk River aquifer, Alberta, Canada: Applied Geochemistry, v. 6, p. 447-464
Faure, G., 1986, Principles of isotope geology: New York, John Wiley & Sons, 589 p.
Ferronsky, V. I., and Polyakov, V. A., 1982, Environmental isotopes in the hydrosphere: John Wiley & Sons, New York, 466 p.
Fish, William, 1993, Chapter 3, Sub-surface redox chemistry--A comparison of equilibrium and reaction-based approaches; in, Metals in Groundwater, H. E. Allen, E. M. Perdue, and D. S. Brown, eds.: Lewis Publishers, Boca Raton, Florida, p. 73-101
Freeze, R. A., and Cherry, J. A., 1979, Groundwater: Prentice-Hall, Inc., Englewood Cliffs, New Jersey, 604 p.
Foster, M. D., 1950, The origin of high sodium bicarbonate waters in the Atlantic and Gulf Coastal Plains: Geochimica et Cosmochimica Acta, v. 1, p. 33-48
Fournier, R. O. and Potter, II, R. W., 1982, An equation correlating the solubility of quartz in water from 25°C to 900°C at pressures up to 10,000 bars: Geochimica et Cosmochimica Acta, v. 46, p. 1,969-1,973
Garneau, G. W., 1995, Detection and characterization of the distribution of mineral intrusion in the Great Bend Prairie aquifer, south-central Kansas: M.S. thesis, The University of Kansas, Lawrence. 102 p.
Garrels, R. M., and Christ, C. L., 1965, Solutions, minerals, and equilibria: San Francisco, California, Freeman, Cooper & Company, 450 p.
Garrels, R. M., and Mackenzie, F. T., 1971, Evolution of sedimentary rocks: New York, W. W. Norton, 397 p.
Geiger, C. O., Lacock, D. L., Schneider, D. R., Carlson, M. D., and Dague, B. J., 1994, Water resources data Kansas water year 1994: U.S. Geological Survey, Water-data Report KS-94-1, p. 469-474
Gerritse, R. G., and Adeney, J. A., 1992, Tracers in recharge--Effects of partitioning in soils: Journal of Hydrology, v. 131, p. 255-268
Heck, B. A., Myers, N. C; and Hargadine, D. A., 1992, Hydrogeology and ground-water-quality conditions at the Reno County landfill, south-central Kansas, 1990-91: U. S. Geological Survey, Water-resources Investigations Report 92-4169,56 p. [available online]
Hem, J. D., 1985, Study and interpretation of the chemical characteristics of natural water: U. S. Geological Survey, Water-supply Paper 2254, 263 p. [available online]
Hendry, M. J., Gillham, R. W., and Cherry, J. A., 1983, An integrated approach to hydrogeologic investigations--A case history: Journal of Hydrology, v, 63, p. 211-233
Holser, W. T., 1979, Trace elements and isotopes in evaporites; in, Marine Minerals, R. G. Burns, ed.: Mineralogical Society of America, Short Course Notes 6, p. 295-346
Johnson, R. L., Keely, J. F., Palmer, C. D., Suflita, J. M., and Fish, W., 1989, Transport and fate of contaminants in the subsurface: Environmental Protection Agency, Seminar Publication no. EPA/625/4-89/019, 148 p.
Klein, c., and Hurlbut, Jr., C. S., 1993, Manual of mineralogy, 21st edition: New York, John Wiley & Sons, 681 p.
Kororn, S. F., 1992, Natural denitrification in the saturated zone--A review: Water Resources Research, v. 28, no. 6, p. 1,657-1,668
Kukuk, M. S., 1987, Effects of the Barton County landfill on local water quality: Kansas Geological Survey, Open-file Report 87-8, 93 p.
Kung, K. J. S., 1990, Preferential flow in a sandy vadose zone, 1. Field observation: Geoderma, v. 46, p. 51-58
Land, L. S., 1995a, Na-Ca-Cl saline formation waters, Frio Formation (Oligocene), south Texas, USA--Products of diagenesis: Geochimica et Cosmochimica Acta, v. 59, no. 11, p. 2,163-2,174
Land, L. S., 1995b, The role of saline formation water in crustal recycling: Aquatic Chemistry, v. 1, no. 2, p. 137-145
Long, D. T., and Angino, E. E., 1982, The mobilization of selected trace metals from shales by aqueous solutionsEffects of temperature and ionic strength: Economic Geology, v. 77, no. 3, p.646-652
Mabey, W. R., et al., 1982, Aquatic fate process data for organic priority pollutants: U.S. Environmental Protection Agency, Chapter 4, EPA/440/4-81-014
Macfarlane, P. A., Whittemore, D.O., Townsend, M. A., Doveton, J. H., Hamilton, V. J., Coyle, W. G., III, Wade, A., Macpherson, G. L., and Black, R. D., 1989, The Dakota Aquifer Program--Annual Report, FY89: Kansas Geological Survey, Open-file Report 90-27, 301 p. [available online]
Macpherson, G. L., 1994, Bromide content of Pennsylvanian black shales, implications for sources of halogens in formation waters: Geological Society of America, Abstracts with Programs, v. 26, no. 7, p. 164
Macpherson, G. L., McElwee, C. D., Butler, J. J., Jr., and Bohling, G. C., 1996, Water chemistry variation in an alluvial aquifer--Multi-level samplers reveal more than screened wells: Geological Society of America, Abstracts with Programs, v. 28, no. 7, p. 460
Magoon, L. B., and Dow, W. G., 1994, The petroleum system--From source to trap: American Association of Petroleum Geologists, Memoir 60, 655 p.
Matisoff, G., Khourey, C. J., Hall, J. F., Varnes, A. W., and Strain, W. H., 1982, The nature and source of arsenic in northeastern Ohio ground water: Ground Water, v, 20, no. 4, p. 446- 456
Mazor, Emanuel, 1991, Applied chemical and isotopic groundwater hydrology: Open University Press, Buckingham, United Kingdom, 274 p.
Merriam, D. F., 1963, The geologic history of Kansas: Kansas Geological Survey, Bulletin 162, 317 p. [available online]
Miller, R. E., Randtke, S. J., Hathaway, L. R., and Denne, J. E., 1991, A survey of organic carbon and trihalornethane formation potential in Kansas ground waters: Kansas Geological Survey, Chemical Quality Series 12, 30 p. [available online]
Miller, R., and Donahue, R. L., 1990, Soils--An introduction to soils and plant growth, 6th edition: Prentice-Hall, Englewood Cliffs, New Jersey, 750 p.
Milliken, K. L., 1989, Petrography and composition of authigenic feldspars, Oligocene Frio Formation, south Texas: Journal of Sedimentary Petrology, v. 59, no. 3, p. 361-374
Moran, S. R., Geonwold, G. H., and Cherry, J. A., 1978, Geologic, hydrologic, and geochemical concepts and techniques in overburden characterization for mined-land reclamation: North Dakota Geological Survey, Report of Investigation No. 63, 152 p.
Murray, J. W., 1975, The interaction of metal ions at the manganese dioxide-solution interface: Geochimica et Cosmochimica Acta, v. 39, p. 505-520
Myers, N. C., Heck, B. A., and Hargadine, D. A., 1993, Hydrogeology and ground-water-quality conditions at the Sumner County landfill, south-central Kansas, 1989-1990: U. S. Geological Survey, Water-resources Investigation 92-4177, 52p. [available online]
Parker, F. P., and Suarez, D. L., 1990, Irrigation water quality assessments; in, Agricultural Salinity Assessment and Management, K. K. Tanji, ed.: American Society of Civil Engineers, New York, p. 220-236
Parker, R. L., 1967, Data of geochemistry, 6th edition, Chapter D, Composition of the Earth's Crust: U. S. Geological Survey, Professional Paper 440-D, 17 p. [available online]
Parkhurst, D. L., Thorstenson, D. C. and Plummer, L. N., 1990, PHREEQE--a computer program for geochemical calculations: U.S. Geological Survey, Water-resources Investigations 80-96, 193 p. [available online]
Pearson, F. J., Jr., Balderer, W., Loosli, H. H., Lehmann, B. E., Matter, A., Peters, T., Schmassmann, H., and Gautschi, A., 1991, Applied isotope hydrogeology--a case study in northern Switzerland: Amsterdam, Elsevier, 439 p.
Perry, C. A., and Anderson, M. R., 1991, Statistical comparison of selected chemical constituents in water from chemigation and conventional irrigation wells in Kansas, 1987: U.S. Geological Survey, Water Resources Investigations Report 91-4049 [available online]
Piper, A. M., 1944, A graphic procedure in the geochemical interpretation of water-analysis: Transactions, American Geophysical Union, p. 914-923
Pipkin, B. W., 1994, Geology and the environment: New York, West Publishing Company, 476 p.
Pope, L. M., 1995, Atrazine in surface water and relation to hydrologic conditions within the Delaware River basin pesticide management area, northeast Kansas, July 1992 through December 1994: U.S. Geological Survey, Fact Sheet FS-196-95
Pope, L. M., Brewer, L. S., Foley, G. A., and Morgan, S. C., 1996, Concentrations and transport of atrazine in the Delaware River-Perry Lake system, northeast Kansas, July 1992 through September 1995: U.S. Geological Survey, Open-file Report 96-331,142 p.
Postma, D., Boesen C., Kristiansen, H., and Larsen, F., 1991, Nitrate reduction in an unconfined sandy aquifer-Water chemistry, reduction processes, and geochemical modelling: Water Resources Research, v. 27, p. 2,027-2,045
Rasmussen, P. P., Shockley, J. c., and Hargadine, D. A., 1994, Hydrogeology and water-quality conditions at the City of Olathe landfill, east-central Kansas, 1990-93: U. S. Geological Survey, Water-resources Investigations Report 94-4166, 44 p. [available online]
Robertson, F. N., 1975, Hexavalent chromium in ground water in Paradise Valley, Arizona: Ground Water, v. 13, no. 6, p. 516-527
Siever, R., 1962, Silica solubility, 1-200°C, and the diagenesis of siliceous sediments: Journal of Geology, v. 70, p. 127-150
Simpkins, W. W., and Parkin, T. B., 1993, Hydrogeology and redox geochemistry of CH4 in a late Wisconsinan till and loess sequence in central Iowa: Water Resources Research, v. 29, p. 3,643-3,657
Smith, R. L., Howes, B. L. and Duff, J. H., 1991, Denitrification in nitrate-contaminated ground water--Occurrence in steep vertical geochemical gradients: Geochimica et Cosmochimica Acta, v. 55, p. 1,815-1,825
Sophocleous, M., Townsend, M. A., Orountiotis, C., Evenson, R. A., Whittemore, D.O., Watts, C. E., and Marks, E. T., 1990, Movement and aquifer contamination potential of atrazine and inorganic chemicals in central Kansas croplands: Kansas Geological Survey, Ground Water Series 12, 64 p. [available online]
Sposito, G., 1989, The chemistry of soils: New York, Oxford University Press, 277 p.
Squillace, P. J., Thurman, E. M., and Furlong, E. T., 1993, Ground water as a nonpoint source of atrazine and deethylatrazine in a river during base flow conditions: Water Resources Research, v. 29, no. 6, p. 1,719-1,729
Stamer, J. K., Gunderson, K. D., and Ryan, B. J., 1995, Atrazine concentrations in the Delaware River, Kansas: U.S. Geological Survey, Fact Sheet FS-00l94
Steichen, J., Koelliker, J., Grosh, D., Heiman, A., Yearout, R., and Robbins, V., 1988, Contamination of farmstead wells by pesticides, volatile organics, and inorganic chemicals in Kansas: Ground Water Monitoring Review, v. 8, no. 3, p. 153-160
Stumm, W., and Morgan, J. J., 1981, Aquatic chemistry, 2nd edition: New York, John Wiley & Sons, 780 p.
Tanji, K. K., 1990, Nature and extent of agricultural salinity; in, Agricultural Salinity Assessment and Management, K. K. Tanji, ed.: American Society of Civil Engineers, New York, p.I-17
Thrailkill, J., 1968, Chemical and hydrologic factors in the excavation of limestone caves: Geological Society of America, Bulletin, v. 79, p. 19-46
Thurman, E. M., Goolsby, D. A., Meyer, M. T., Mills, M. S., Pomes, M. L., and Kolpin, D. W., 1992, A reconnaissance study of herbicides and their metabolites in surface water of the midwestern United States using immunoassay and gas chromatography/mass spectrometry: Environmental Science and Technology, v. 26, no. 12, p. 2,440-2,447
Townsend, M. A., 1995, Potential sources of nitrate in south fork of Beaver Creek watershed, Sherman County, Kansas: Kansas Geological Survey, Open-file Report 95-73, 25 p.
Townsend, M. A., and Sleezer, R. O., 1994, Factors affecting nitrate concentration in ground water in south-central Kansas: American Society of Agronomy, 1994 Annual Meeting Abstracts, November 13-18, 1994, Seattle, Washington, p. 48
Townsend, M. A., and Sleezer, R. O., 1995, U.S. Environmental Protection Agency Pollution Prevention Demonstration Project Harvey County, Kansas, Final Report: Kansas Geological Survey, Open-file Report 95-23, 143 p.
Townsend, M. A., Young, D. P., and Healey, J., 1997, Results of agrichemical survey of ground water in Kansas, 1993-1994: Kansas Geological Survey, Open-file Report 98-22, 47 p.
van Riernsdijk, W. H., and Heimstra, T., 1993, Adsorption to heterogeneous surfaces; in, Metals in Ground Water, H. E. Allen, E. M. Perdue, and D. S. Brown, eds: Boca Raton, Florida, Lewis Publishers, p. 1-36
Waples, D. W., 1980, Time and temperature in petroleum formation--Application of Lopatin's method to petroleum exploration: American Association of Petroleum Geologists, Bulletin, v. 64, no. 6, p. 916-926
White, W., B., 1988, Geomorphology and hydrology of karst terrains: Oxford University Press, New York, 464 p.
Whittemore, D.O., 1993, Ground-water geochemistry in the mineral intrusion area of Groundwater Management District No. 5, south-central Kansas: Kansas Geological Survey, Open-file Report 93-2, 110 p.
Whittemore, D.O., 1995a, Geochemical differentiation of oil and gas brine from other saltwater sources contaminating water resources--Case studies from Kansas and Oklahoma: Environmental Geosciences, v. 2., no. 1, p. 15-31
Whittemore, D.O., 1995b, Upper Arkansas River corridor study--Problem identification, preliminary research, and FY96 and FY97 plans: Kansas Geological Survey, Open-file Report 95-70, 30 p.
Whittemore, D.O., and Langmuir, D., 1975, The solubility of ferric oxyhydroxides in natural waters: Ground Water, v. 13, no. 4, p. 360-365
Young, D. P., 1995, Effects of ground-water pumpage on freshwater-saltwater transition zone characteristics, water quality, and water levels at the Siefkes intensive study site, Stafford County, Kansas: Kansas Geological Survey, Open-file Report 95-45c, 23 p.
Previous--Is Sustainability a Viable Concept in the Management of Confined Aquifers in Kansas? || Next--Yield Estimates for Surface-water Sources
Kansas Geological Survey
Comments to webadmin@kgs.ku.edu
Web version placed online May 27, 2013. Original publication date 1998.
URL=http://www.kgs.ku.edu/Publications/Bulletins/239/Macpherson/index.html