Kansas Geological Survey, Bulletin 119, Part 3, originally published in 1956

Originally published in 1956 as Kansas Geological Survey Bulletin 119, Part 3. This is, in general, the original text as published. The information has not been updated.
Results of a study of the composition of 325 samples of limestone from the eastern third of Kansas are presented. Included are chemical analyses for the major constituents of all samples and spectrographic analyses of the minor constituents of 288 of the samples. Special emphasis is placed on the suitability of the limestones for various industrial and agricultural uses, depending upon their composition.
No simple correlation is found between composition and geologic or geographic position. An irregular increase in calcium carbonate content southwestward along the outcrop of a given bed, and also southeastward in a direction normal to the line of outcrop, from younger to older beds, does, however, indicate a general tendency in variation of composition. Some ability to predict composition of samples within small areas is foreseen.
This study was first conceived in 1946 concurrent with the organization of the geochemistry division of the Geological Survey. The study did not gain any momentum until 1949, but from that date until the fall of 1955 collecting and analysis of samples proceeded steadily.
The primary aims of the study are two. First, the State Geological Survey needed the broad coverage of data on limestone that is necessary for an agency responsible for development of mineral resources. Secondly, data on composition would be available for references from the beginning of the study and would subsequently form the logical basis for more complex studies.
Interest in the composition of limestone centers about two diametrically different points of view. From the point of view of the industrial user, a high calcium limestone is required. In the production of quick and hydrated lime, whiting, and white cement, the presence of iron, manganese, or vanadium is undesirable. In ordinary cement the amounts of phosphorus, magnesium, and titanium are of concern. In the manufacture of acetylene, a high calcium stone is demanded that is low in aluminum, phosphorus, arsenic, antimony, and sulfur. For any metallurgical use limestone must be virtually pure calcium carbonate containing only minute percentages of silicon, phosphorus, and sulfur. For complete specifications on limestone for various industrial uses the reader is referred to Kansas Geological Survey Bulletin 90, Part 5 (Runnels, 1951) or references dealing directly with a specific use.
The other point of view is held by agricultural consumers of limestone. Since about 1935 the use of crushed limestone on soils has become common practice. The federal subsidy system, which promoted the use of crushed limestone on soil, acquainted farmers with the beneficial results of this treatment. It has become evident also, that, in addition to calcium carbonate, the small amounts of many other elements usually present in limestone were beneficial to soils and plants. There are some elements that are known to be detrimental to soils and plants. Soil scientists and agronomists (McMurtrey and Robinson, 1938) have compiled a list of known helpful and harmful elements. The elements usually regarded as beneficial, when present in the top 6 to 8 inches of soil in small amounts, are boron, zinc, copper, manganese, and molybdenum. Boron is needed in amounts of 1 to 2 pounds of available boron per acre. Some plants require soil containing zinc in a concentration of 2 1/2 pounds per acre. Copper is beneficial when present in soils in the amount of 2 pounds per acre, but addition of as little as 2 ounces per acre will greatly benefit depleted soils. The proper amount of manganese is believed to be 25 to 50 pounds per acre, or sufficient to produce 1 1/2 to 2 1/2 pounds of available, manganese per acre. The presence of approximately one ounce of molybdenum per acre has been found to greatly stimulate the process of nitrogen fixation. The elements known to be deleterious to plant life, and, just as important, harmful to animal consumers of the plants when present in excessive amounts are silver, selenium, tellurium, thallium, arsenic, antimony, chromium, and lead. From the standpoint of use as agricultural limestone, it was evident that knowledge of the composition of Kansas limestones would not be complete without investigation of the minor elements in these stones.
A third reason for a study of the composition of Kansas limestones is that a basic scientific investigation is of long-range value. Knowledge of the variation of the major constituents might be applied to studies of stratigraphy and environment of deposition. The minor constituents might be of aid in geochemical prospecting and geochemical and stratigraphic studies.
This study reports the composition of 325 samples of Kansas limestone. It encompasses the limestone of approximately the eastern one-third of Kansas. The composition was determined by both chemical and spectrochemical methods and thus the study covers both major and minor constituents. The geologic age of the rocks that were sampled ranges from Mississippian through Pennsylvanian into Early Permian (Fig. 1).
Figure 1--Index map of eastern Kansas showing localities where samples were colected (generalized).
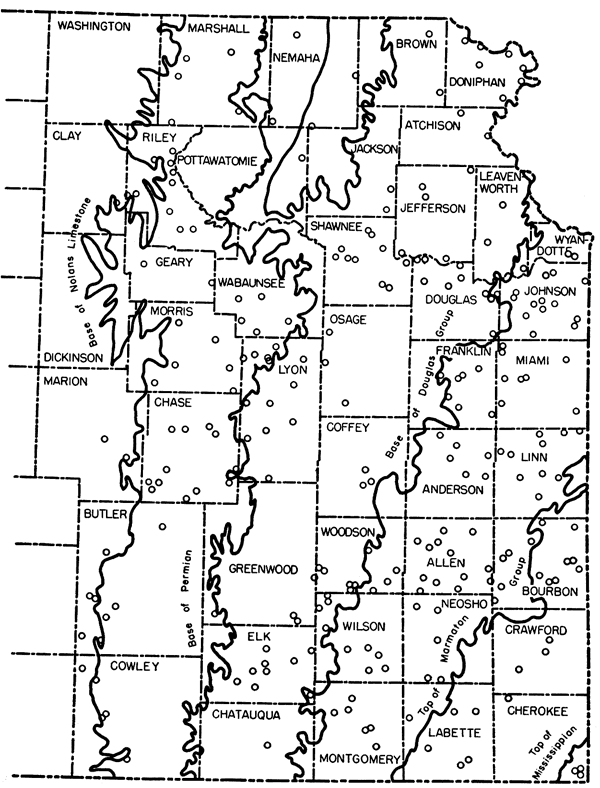
No attempt has been made in this study to show the field relations or to discuss the stratigraphy of the limestones sampled. Complete notes were taken and references were made to both published and unpublished work by the various geologists on the survey staff. All correlations and nomenclature are from Kansas Geological Survey Bulletin 89 (Moore and others, 1951).
Special thanks should be given to members of the Survey who collected samples and advised on stratigraphy: Robert O. Kulstad collected from 1949 through 1951. William Ives collected during the summers of 1952 and 1953. Dr. J. M. Jewett gave invaluable advice on stratigraphic identification throughout the study. Analysts include E. E. McGill, H. S. Van Nortwick, Lois M. Lloyd, and Loretta Vorse.
We also wish to thank all the property owners and operators, whose exceptional cooperation helped us to a very great extent. Many County Engineers also assisted by suggesting new locations for sampling.
There are many factors to be considered in undertaking a project covering a large area where the rocks comprise numerous formations. The initial decision was to collect from any deposit where there is a minimum thickness of 3 feet or which had been worked commercially. Many of the old quarries were located by the use of the Survey publication Kansas Pits and Quarries (Kulstad and Nixon, 1951). In a few places limestone beds less than 3 feet thick were sampled. Most of these were very persistent beds.
The samples taken weighed 6 to 25 pounds each. They consisted of chips from a continuous surface from top to bottom of the ledge, taken from a fresh surface if possible. In a few places drill dust was collected. Stratigraphic and geographic locations were determined and recorded in field notes.
The samples from the field were first crushed in a jaw crusher, then put through a gyratory crusher, and finally passed through a roll mill several times. After this reduction to a coarse powder the samples were split repeatedly with a Jones riffle-splitter until each half was about 100 grams. One portion was preserved for future reference. The other portion was then ground with a mortar and pestle, either by hand or with a Fisher Improved mortar grinder, until 100 percent of the material passed an 80-mesh screen. The thoroughly mixed sample was retained for analysis.
The procedures used for all major constituents are ones used for nonmetallic mineral analysis. The various methods used were selected (cf. Hillebrand and Lundell, 1929, 1946; Kolthoff and Sandell, 1943, 1946; Scott, 1939; Willard and Diehl, 1943, 1946) to give acceptable accuracy without consuming an unreasonable length of time. A brief description of these methods follows.
Loss on ignition--The approximate carbon dioxide content of a sample may be determined by the weight loss on ignition at 1000° C. Samples that are very impure will lose water of hydration in addition to carbon dioxide, whereas for high-calcium samples the carbon dioxide will be the only loss. This determination was made using a temperature-controlled laboratory muffle. The samples were preheated at 400° C for 20 minutes to remove sulfide sulfur, if present, then heated at 1000° C for 40 to 60 minutes. The net loss of weight was recorded as loss on ignition.
Silica--Silica was separated by dehydration with perchloric acid. The same sample used for loss on ignition was used for all subsequent determinations except sulfide sulfur, sulfate sulfur, sodium, potassium, and phosphorus. Dehydration was accomplished by refluxing the solution for 20 minutes. The crude silica was filtered, washed, and ignited to 1200° C in platinum crucibles. The ignited and weighed crude silica was treated with sulfuric and hydrofluoric acids to volatilize the silica as silicon tetrafluoride. The residue was ignited to 1000° C and weighed. The loss of weight represented total silica.
R2O3 group--The residue from the crude silica was combined with the filtrate from the silica determination. The combined portions were boiled with a few drops of bromine water, then precipitated with ammonium hydroxide at a pH of 6. If the precipitate was large a second precipitation was made. The filtered and dried precipitate was ignited in porcelain crucibles to 1200° C and weighed. This precipitate consisted of alumina, phosphorus pentoxide, and ferric oxide in all samples, and manganese dioxide, titania, and vanadium pentoxide if present. This precipitate was used to determine ferric oxide, after suitable fusion and solution, by titration with potassium dichromate. After separate determination of phosphorus pentoxide, the alumina was obtained by difference. Titania, manganese dioxide, and vanadium pentoxide are reported as alumina.
Calcium oxide--Calcium was precipitated from the R2O3 filtrate by double precipitation with ammonium oxalate followed by titration with ceric sulfate. The combined filtrates from this precipitation were used for determination of magnesium after destruction of ammonium and oxalate compounds with concentrated nitric and hydrochloric acids.
Magnesium oxide--The residue from the calcium determination was used for determining magnesium. The method used was the double precipitation of magnesium ammonium phosphate. The final precipitate was ignited to 1100° C, cooled, and weighed.
Phosphorus pentoxide--This determination was made by treating a 3-gm sample with 20 percent nitric acid, filtering through a retentive paper, then precipitating the phosphorus with ammonium citromolybdate solution. The canary-yellow precipitate was filtered on fritted glass crucibles, dried at 105 to 110° C, and weighed. When care is used to obtain a stable complex this method is accurate and very fast.
Sulfate sulfur--A 3-gm sample was treated with 10 percent hydrochloric acid, diluted to 100 or 150 cc with hot water, and filtered. The filtrate contained the sulfate present in the original sample and was precipitated with barium chloride from a dilute solution. The resultant barium sulfate was then filtered, ignited, reoxidized, reheated, cooled, and weighed.
Sulfide sulfur--This determination actually is total sulfur, sulfide sulfur being calculated by difference between total sulfur and sulfate sulfur. A mixture of bromine and carbon tetrachloride was used to oxidize all sulfide (or elemental) sulfur to the sulfate form. The procedure for sulfate sulfur was then followed to determine total sulfur. The difference between this value and the value for sulfate sulfur represents sulfide sulfur expressed as sulfur trioxide. This can then be reported as such or calculated to S.
Sodium and potassium oxides--This determination was made only on a selected group of samples. Most limestones contain only a small amount of alkalis. If the summation of the other determinations showed more than traces of these elements, they were determined separately. The determinations were made using a Perkin Elmer model 52-C flame photometer.
Tables 1 to 29 show the results of the analyses for all constituents. Calculated values are shown for calcium carbonate, magnesium carbonate, and calcium carbonate equivalent. This last figure is the total carbonate content computed as calcium carbonate. Where magnesium is present in relatively large amounts this calculation will show a higher equivalent value because magnesium has a lower molecular weight than calcium. Conversely, if much iron carbonate is present this same calculation would give a smaller equivalent figure than the sum of calcium plus iron carbonates. The calculation of calcium carbonate equivalent can be made by using the loss-on-ignition value or by using the sum of the carbon dioxide contents of the calculated calcium and magnesium carbonates. Either method can be slightly in error, because small errors in the analysis are magnified. Both methods were used in preparing this calculated value.
The remainder of the 100-gram sample retained for chemical analysis was remixed to eliminate possible segregation by particle size, and about 1 gram removed for spectrographic analysis of trace elements. The 1-gram sample was ground in a 55-mm mullite mortar until 100 percent would pass a 200-mesh sieve, and then weighed. A matrix of anhydrous lithium sulfate was added in the ratio of one part of lithium sulfate to two parts of the ground limestone, and the mixture reground until homogeneous. The finely ground lithium salt, although anhydrous when added to the limestone, absorbed some moisture from the air during regrinding. This caused no change in appearance, behavior, or grinding characteristic, but did tend to cause the mixture to stick to the fine sieve, and therefore the mixture was not resized after grinding.
The samples were stored, prior to analysis, in small glass vials under bakelite screw-type caps having plastic liners.
The standards used were prepared from a matrix of Specpure Grade (Johnson, Matthey & Company Ltd.) calcium carbonate. To this matrix was added 0.005 percent each of Mn, Zn, B, Mo, Ag, and Pb, 0.0005 Ni, 0.03 Cr, and 0.2 percent Ba, for the most part in the form of the pure oxide. This base standard was then diluted with an equal amount of CaCO3 to make a second standard, the second standard was diluted with four parts of CaCO3 to make a third standard, and the third standard diluted with nine parts of CaCO3 to make a fourth standard. A standard for higher concentrations of Mn was made in a similar manner, starting with an initial concentration of 0.500 percent Mn, and successively diluting until standards were available in the concentrations of 0.500, 0.250, 0.050, and 0.025 percent.
The special standards for vanadium were prepared, in four steps, to contain 0.100, 0.025, 0.010, and 0.0025 percent of V, added as ammonium vanadate.
The standards for copper were somewhat more difficult to prepare. The copper concentration in some of the limestone samples was found to be appreciably lower than the concentration in the combined calcium carbonate-lithium sulfate matrix mixture. The smallest amount of copper present in any of the samples collected in the early part of the study was that in sample 50-202, a sample of Altamont limestone from Crawford County. By using special purity electrodes and by use of the successive-addition method the true concentration of copper in this limestone was determined to be 18 X 10-6 percent. The standards prepared for the successive-addition determination were then of a known concentration, and were used as standards for the determination of the copper concentration in all the other unknowns. These standards were x percent, x + 7 X 10-6 percent, and x + 23 X 10-5 percent, which were found to be 18 X 10-6 percent, 25 X 10-6 percent, and 25 X 10-5 percent, respectively. Assuming x to be negligible in the presence of large amounts of added copper, standards containing 0.0025, 0.010, and 0.025 percent were prepared from sample 50-202 and used. These last three standards were rarely of value, and were used only for samples that proved upon analysis to be above the range of the first three standards.
The spectrograph used for these analyses was an Applied Research Laboratories 1.5-meter grating-type instrument equipped with a D.C. Arc source, and using 35-mm film as the recording medium. The optical arrangement permits utilization of the ultra-violet spectrum from 2200Å to 4200Å.
The electrodes were shaped from 0.242-inch standard purity graphite rod in the form of an undercut concave platform with a centerpost extending from the center of the concavity to a height of about 1.6 mm above the rim of the platform. The counterelectrodes were shaped in the form of a conical point of much the same angle as a pencil point. This shape has the disadvantage of burning away somewhat rapidly and thus not maintaining a constant analytical gap, but the advantage of extreme localization of the arc by the sharp point to the centerpost of the sample electrode outweighed the disadvantage. Furthermore the rate of change of the analytical gap can be presumed to be nearly constant for both samples and standards if other variables in excitation conditions are held to a minimum.
The sample electrodes were filled on a volume basis to the level of the top of the concave platform. The weight of sample contained in the platform is of no consequence, provided it is about the same for all samples and standards.
The average amount of mixture of sample and lithium sulfate contained upon the platform, observed on about 25 randomly selected filled electrodes, was found to be 14 mg, with a variance of about 7 percent.
The external-standard method gave required accuracy using the standards described above. This method is as follows:
The spectra of four standards and five unknown samples were photographed on each film, to eliminate, insofar as possible, differences in developer strength, temperature, etc. The main inaccuracy of the external-standard method is that there is no sure check or compensation in each spectrum for uncontrollable variables such as arc wandering and differential volatility of unknown elements with variation of matrix composition. A sort of control was established for arc wandering by discarding any spectrum that did not have the same general overall appearance to the eye as the rest of the spectra (standards included). The differential volatility effect was minimized in some measure by the addition of the lithium sulfate. Experimental evidence (Harvey, 1950) shows that the overall deviation from accuracy of this method is 20 to 50 percent of the observed amount for each element. The lower figure is judged to represent the average accuracy obtained in this study.
Four different sets of excitation conditions were required for proper evaluation of each sample, and no satisfactory compromise conditions were found. One exposure sufficed for most of the elements present, but vanadium was too insensitive for this general exposure, copper was too sensitive, even in the minute amounts present, and manganese, except in a few samples, was present in too great an amount to be determined with the rest of the trace elements. The four sets of excitation conditions used are detailed in Table A.
Table A--Excitation conditions used in spectrographic analyses.
| B, Zn, Ni, Mo, Cr, Ag, Ba, Pb, Sr, Ti, and small amounts Mn |
Cu | V | Mn | |
|---|---|---|---|---|
| Current | 5.2a | 5.2a | 5.2a | 5.2a |
| Sector | 30 percent | 15 percent | 50 percent | 30 percent |
| Grating doors | open | open | open | open |
| Gap | 3 mm | 3 mm | 3 mm | 3 mm |
| Time | 60 sec | 30 sec | 45 sec | 30 sec |
| Primary slit | 25µ | 25µ | 20µ | 20µ |
Tables 1 to 29 give the results of the analyses for all constituents.
Some of the samples were not analyzed spectrographically. These were, in general, those samples that had the external appearance of limestone but were found to be calcareous shales. A few samples of this type were found although the locations indicated them to be in the proper stratigraphic positions for normal limestones.
The total number of samples analyzed spectrographically was 288 as compared with 325 reported for major constituents. The elements reported from this spectrographic procedure are zinc, boron, barium, nickel, molybdenum, vanadium, titanium, manganese, lead, chromium, silver, and copper. Selected samples were analyzed for strontium. All other metallic elements were found to be absent or present in such minute amounts as to be undetectable.
The limestones analyzed showed a marked diversity of composition, both in the major constituents and the amount and numbers of trace elements present. The purity, which for this study is defined as the percent of calcium carbonate present, ranges from a maximum of 98.38 percent to a minimum of 22.15 percent, and averages 89.79 percent. The limestones in the lower range are hardly more than calcareous shales, although found in the position of limestones, stratigraphically. The purity seems to increase in direct proportion to the age of deposition. That increase is from northwest to southeast in the state, and from younger to older across the various stratigraphic units. It must be realized, however, that no statements of such a general nature can be taken as applying specifically to any one area or outcrop. Continually we found samples that not only followed no rule, but in fact seemed to be completely contrary to the general trend of similar samples. As one example of this, eleven samples were taken of the Raytown limestone, a member of the Iola formation, Kansas City group. Of these eleven, eight contained more than 90 percent calcium carbonate, two were between 80 and 90 percent, but the other sample contained the minimum of 22.15 percent calcium carbonate.
In this same manner, many of the limestone members showed one or more anomalous high or low results as compared with other samples in the same member in other, nearby areas. It was not predictable which of the other major constituents would increase or decrease with the fluctuation of the calcium carbonate content. Some of the samples that did not conform to expectations were, in fact, collected from localities very near to others that gave predictable results.
The Hartford limestone, a member of the Topeka formation, Shawnee group, yielded a sample containing 28.33 percent magnesium carbonate. A sample from the same member at a locality less than a mile away contained only 1.11 percent, conforming to the other four samples of this limestone, which ranged from 1.07 to 6.29 percent. The Hartford, of course, is a relatively thin limestone, being only 5 feet thick or less at all locations sampled. A thicker limestone, the Towanda member of the Doyle formation, Chase group, shows the same irregularity of composition. Three of the four samples analyzed contained 1.36, 3.49, and 1.74 percent magnesium carbonate, but the fourth sample, from a locality less than a mile from that of the last mentioned, contained 11.26 percent.
It should be mentioned that the Permian limestones were more magnesian than the Pennsylvanian, and indeed, upon calculation of ratios that consider only the calcium and magnesium carbonates, some of the Permian samples were found to contain the two carbonates in very nearly the ratio of 1.186/1 that is characteristic of dolomite.
The Nolans formation, represented by the Herington and Krider members, showed an unusual feature. The three samples collected in Dickinson County were relatively impure limestones and contained only small amounts of magnesium carbonate (2 to 6 percent). All other samples from this formation, collected at locations outside of Dickinson County, contained 30 to 40 percent magnesium carbonate, and were, in fact, essentially impure dolomite. The average magnesium carbonate concentration for all limestones analyzed, even including those very high values in the Permian, fell slightly below the average of large groups of limestone analyses reported elsewhere. Two groups of 345 and 498 samples respectively, analyzed by the U.S. Geological Survey, averaged 7.9 and 4.5 percent magnesium oxide (Clarke, 1924), whereas our average was 1.80 percent. As might be expected, the higher magnesium content of the 843 samples analyzed by the U.S. Geological Survey is accompanied by a lower amount of CaO. The two groups averaged 42.6 and 40.6 percent CaO (76.1 and 72.5 percent CaCO3) respectively, whereas the average of the Kansas limestones described in this report was 48.5 percent CaO (86.6 percent CaCO3).
The other major elements found in the Kansas limestones can be summarized as follows:
Silica (SiO2) ranged from a low value of 0.16 percent to a maximum of 66.84 percent (although only a very few samples even approached this high value), and averaged 6.75 percent.
The alumina, the percentage of which as reported here also includes the oxides of manganese, titanium, and vanadium, if present, ranged from 0.08 to 8.89 percent, averaging 1.27 percent.
The Fe2O3 content attained a minimum of 0.10 percent and a maximum of 7.03 percent, and averaged 1.32 percent. The alumina analyses seemed to agree with the analyses made by the U.S. Geological Survey, but the Fe2O3 content was considerably greater in the Kansas samples than the averages given by the U.S. Geological Survey, 0.54 and 0.77 percent.
It seemed, as these maxima, minima, and averages were being calculated, that perhaps the figures would be misleading, for no allowance is made for the thickness of the ledges of stone. Under the system of straight arithmetical averages, a thick bed of high-calcium limestone is given no greater significance than a thin bed of low-calcium limestone. The reverse, of course, is also true. Consideration of this fact led us to recalculate the averages weighted by the thickness of each bed sampled. These weighted averages were obtained in the following manner. The percentage of each major element in a sample was multiplied by the thickness of the rock at the point where sampled. The resulting feet-percent for each element were added together for all the samples, and the total feet-percent divided by the total thickness of all the samples represented. The arithmetical and weighted averages compare as follows:
| Arithmetical | Weighted | |
|---|---|---|
| CaCO3 | 86.6 percent | 87.89 percent |
| MgCO3 | 3.76 | 3.39 |
| SiO2 | 6.75 | 6.09 |
| Al2O3 | 1.27 | 1.16 |
| Fe2O3 | 1.32 | 1.14 |
It is seen that the weighted average is somewhat higher in calcium carbonate and somewhat lower in the other major constituents than the arithmetical average. Therefore, the thicker limestones, which by the nature of the calculation affect the weighted average more than do the thinner ones, seem to show a somewhat higher calcium carbonate content--a greater purity--than do the thinner ones.
The minor constituents showed a trend exactly the reverse of that found in the major constituents. As the purity of the limestones increased, that is, progressively down the stratigraphic column, the minor elements, as determined spectrographically, showed a tendency to increase. Toward the southeast portion of the state, more of the minor elements showed higher maxima than in any other area. The maxima of all the minor elements were spotty, the higher concentrations usually being grouped in certain areas, but in many places each high value was surrounded by an area of much lower concentration.
One area found to be low in trace elements both qualitatively and quantitatively was Chase County and small portions of the counties surrounding it. The low concentration in this large area is as yet unexplained.
An attempt was made to correlate concentration of minor elements with some other factor, geologic or geographic. This attempt did not meet with success. The results of the analyses showed a partial correlation with topographic position, stratigraphic unit, and geographic location, but each of these seemed to affect the other, and no way of arranging these complex factors was found that would present a true picture of correlation, as the effect of each factor singly was undeterminable. The amount and number of trace elements in any given ledge of rock, however, did seem to increase in a southerly direction, so that any ledge of rock was generally greater in both calcium carbonate and trace element content and of lower content in other major constituents towards its southern end as compared to the northernmost exposures. In many beds this increase from north to south was greater than the aforementioned increase of purity with increasing age of deposition. That is, any given ledge of rock might, toward its southern end, contain more calcium carbonate and trace elements and less other major constituents than the northerly outcrops of an older and lower ledge of limestone. We were unable to note any general change in major or minor constituents in the vicinity of the two intrusive domes in Woodson County or the five small intrusive pipes in Riley County. Nor was there any marked change in the lead or zinc content of the limestones as one progressed toward the Tri-state district. The resume of results obtained by spectrographic analysis may be seen in Table B.
Table B--Resume of Spectrographic Results.
| Element | No. of samples containing |
Minimum | Maximum | Average on samples containing |
Average on all samples |
||||
|---|---|---|---|---|---|---|---|---|---|
| % | oz/ton | % | oz/ton | % | oz/ton | % | oz/ton | ||
| B | 95 | 0.00005 | 0.016 | 0.03 | 9.6 | 0.0021 | 0.67 | 0.00069 | 0.22 |
| Cu | 287 | .000013 | .004 | .05 | 16.0 | .0005 | .175 | ||
| Mo | 45 | .00001 | .0032 | .007 | 2.24 | .0044 | 1.41 | .00069 | .22 |
| Mn | 287 | .002 | .64 | .6 | 192 | .085 | 25.38 | ||
| Ag | 102 | .00001 | .0032 | .002 | .64 | .00009 | .031 | .000034 | .011 |
| Pb | 159 | .0001 | .032 | .02 | 6.4 | .0016 | .51 | .00091 | .29 |
| Zn | 21 | .00005 | .016 | .05 | 16.0 | .0035 | 1.12 | .00349 | .112 |
| Sn | 54 | .0001 | .032 | .02 | 6.4 | .0019 | .61 | .00036 | .12 |
| Ti | 281 | .001 | .32 | .6 | 192 | .0205 | 6.56 | .020 | 6.4 |
| V | 271 | .0005 | .16 | .3 | 96 | .0074 | 2.37 | .0069 | 2.2 |
| Cr | 141 | .0001 | .032 | .02 | 6.4 | .0013 | .42 | .00062 | .20 |
| Ni | 266 | .00005 | .016 | .01 | 3.2 | .0010 | .32 | .00094 | .30 |
| Ba | 50 | .001 | .32 | .3 | 96 | .039 | 12.48 | .0068 | 2.2 |
| Sr | 61 only analyzed |
.0014 | .45 | >.20 | >64 | .047 | 15.04 | ||
There seems to be no firm conclusion that can be drawn from the results of the analyses that have been performed, as presented here. The only general trends that were observed were that the calcium carbonate content and the number and amount of trace elements present seem to increase as one proceeds along a given ledge of rock from north to south, or across the ledges of rock from youngest to oldest. Sampling in a line normal to the line of the outcrop, that is, a northwest-southeast direction, is necessary to observe the second trend. Evidence can be found in the tables to show that the most persistent ledges of limestone, particularly the thicker ones, show one or more high-calcium areas, seemingly regardless of their usual or average content. In the main, however, it is possible to make general predictions about the expected composition of an outcropping limestone, if samples from adjacent or surrounding areas have been analyzed. These predictions should be substantiated by check analyses to eliminate the slight possibility that the outcrop in question may be completely anomalous with respect to nearby outcrops. This check analysis can be brief, and could take the form of a determination of loss on ignition. The limestones whose analyses are presented here represent samples from every major limestone ledge cropping out in the eastern third of Kansas.
Clarke, F. W. (1924) The data of geochemistry: U. S. Geol. Survey, Bull. 770, p. 564-580.
Harvey, C. E. (1950) Spectrochemical procedures: Applied Research Laboratories, p. 127-184.
Hillebrand, W. F., and Lundell, G. E. F. (1929, 1946) Applied inorganic analysis: John Wiley and Sons, p. 1-886.
Kolthoff, I. M., and Sandell, E. B. (1943, 1946) Textbook of quantitative inorganic analysis: Macmillan Co., p. 1-765.
Kulstad, R. O., and Nixon, E. K. (1951) Kansas pits and quarries: Kansas Geol. Survey, Bull. 90, pt. 1, p. 1-12. [available online]
McMurtrey, J. E., and Robinson, W. O. (1938) Soils and Men: U. S. Dept. Agriculture yearbook, p. 807-834.
Moore, R. C., Frye, J. C., Jewett, J. M., Lee, Wallace, and O'Connor, H. G. (1951) The Kansas rock column: Kansas Geol. Survey, Bull. 89, p. 1-132. [available online]
Runnels, R. T. (1951) Some high-calcium limestones in Kansas: Kansas Geol. Survey, Bull. 90, pt. 5, p. 1-28. [available online]
Scott, W. W. (ed. by N. H. Furman) (1939) Scott's standard methods of chemical analysis, v. 1, D. Van Nostrand Co., p. 1-1104.
Willard, H. H., and Diehl, H. (1943, 1946) Advanced quantitative analysis: D. Van Nostrand Co., p. 1-421.
| County | Location | Formation | Table No. |
|---|---|---|---|
| Allen | 24S-18E, 19E | Stanton | 18 |
| Allen | 24S-18E, 20E | Iola | 22 |
| Allen | 25S-19E | Iola | 22 |
| Allen | 26S-18E | Iola | 22 |
| Allen | 26S-18E, 19E, 20E | Drum | 23 |
| Allen | 23S, 25S-21E | Dennis | 25 |
| Allen | 26S-20E | Dennis | 25 |
| Allen | 26S-21E | Swope | 26 |
| Anderson | 19S, 20S-19E | Stanton | 18 |
| Anderson | 20S-19E, 21E | Plattsburg | 19 |
| Anderson | 21S-20E | Plattsburg | 19 |
| Anderson | 21S-21E | Iola | 22 |
| Atchison | 5S-19E | Deer Creek | 14 |
| Atchison | 5S, 6S-21E | Oread | 16 |
| Bourbon | 25S, 26S-22E | Dennis | 25 |
| Bourbon | 24S-23E | Swope | 26 |
| Bourbon | 27S-21E | Swope | 26 |
| Bourbon | 23S-23E | Altamont | 28 |
| Bourbon | 23S, 25S-25E | Pawnee | 28 |
| Bourbon | 26S-23E, 25E | Pawnee | 28 |
| Bourbon | 25S-25E | Fort Scott | 28 |
| Brown | 1S-18E | Auburn Shale | 12 |
| Butler | 24S, 25S-4E | Nolans | 1 |
| Butler | 27S, 29S-3E | Nolans | 1 |
| Butler | 27S-3E | Winfield | 2 |
| Butler | 29S-4E | Winfield | 2 |
| Butler | 24S, 26S-6E | Barneston | 4 |
| Butler | 27S, 28S-4E | Barneston | 4 |
| Chase | 22S-6E | Nolans | 1 |
| Chase | 22S-6E, 8E | Winfield | 2 |
| Chase | 21S-6E | Doyle | 3 |
| Chase | 22S-6E, 7E | Barneston | 4 |
| Chase | 18S-9E | Crouse | 8 |
| Chase | 19S-7E, 8E | Beattie | 9 |
| Chase | 22S-9E | Beattie | 9 |
| Chase | 19S-8E, 7E | Eskridge, Grenola, and Red Eagle |
10 |
| Chase | 21S-9E | Foraker | 11 |
| Chautauqua | 33S-11E | Oread | 16 |
| Cherokee | 31S-23E | Cherokee | 29 |
| Cherokee | 34S, 35S-25E | Mississippian | 29 |
| Clay | 9S-4E | Barneston | 4 |
| Coffey | 21S, 22S-15E | Oread | 16 |
| Coffey | 21S-16E | Douglas | 17 |
| Cowley | 31S-3E | Winfield | 2 |
| Cowley | 32S-4E | Barneston | 4 |
| Cowley | 34S-5E | Barneston | 4 |
| Crawford | 27S, 29S-24E | Marmaton | 28 |
| Crawford | 29S-23E | Marmaton | 28 |
| Crawford | 30S-22E | Marmaton | 28 |
| Dickinson | 14S, 16S-4E | Nolans | 1 |
| Doniphan | 1S-19E | Topeka | 13 |
| Doniphan | 2S, 3S-20E | Deer Creek | 14 |
| Doniphan | 2S, 4S, 5S-21E | Oread | 16 |
| Doniphan | 3S-22E | Douglas | 17 |
| Douglas | 12S-17E | Topeka | 13 |
| Douglas | 11S-17E, 18E | Deer Creek | 14 |
| Douglas | 1IS-17E | Lecompton | 15 |
| Douglas | 12S-18E, 19E | Oread | 16 |
| Douglas | 13S-19E | Oread | 16 |
| Douglas | 14S-18E, 20E | Oread | 16 |
| Douglas | 13S-21E | Stanton | 18 |
| Elk | 30S, 31S-10E | Wabaunsee | 12 |
| Elk | 30S-11E | Wabaunsee | 12 |
| Elk | 30S-11E | Deer Creek | 14 |
| Elk | 31S-10E | Deer Creek | 14 |
| Elk | 30S-11E | Lecompton | 15 |
| Elk | 29S-13E | Oread | 16 |
| Elk | 30S-12E | Oread | 16 |
| Elk | 31S-13E | Douglas | 17 |
| Franklin | 16S, 17S-20E | Stanton | 18 |
| Franklin | 18S-19E, 21E | Stanton | 18 |
| Franklin | 17S-19E, 20E | Stanton | 18 |
| Franklin | 17S-19E | Plattsburg | 19 |
| Geary | 12S-5E | Barneston | 4 |
| Geary | 13S-8E | Barneston | 4 |
| Greenwood | 27S-9E | Foraker | 11 |
| Greenwood | 28S-12E | Oread | 16 |
| Greenwood | 26S-13E | Oread | 16 |
| Jackson | 6S-12E | Beattie | 9 |
| Jackson | 6S-12E | Grenola | 10 |
| Jackson | 8S-16E | Wabaunsee | 12 |
| Jefferson | 8S-18E | Topeka | 13 |
| Jefferson | 9S-18E | Deer Creek | 14 |
| Jefferson | 7S-20E | Deer Creek | 14 |
| Jefferson | 11S-17E | Oread | 16 |
| Johnson | 14S-22E, 23E | Stanton | 18 |
| Johnson | 12S-24E | Stanton | 18 |
| Johnson | 13S, 15S-23E | Plattsburg | 19 |
| Johnson | 13S-22E, 23E, 24E | Wyandotte | 20 |
| Johnson | 14S-25E | Wyandotte | 20 |
| Johnson | 12S-25E | Drum | 23 |
| Johnson | 12S-25E | Cherryvale | 24 |
| Labette | 32S-17E | Swope | 26 |
| Labette | 32S-19E, 20E | Marmaton | 28 |
| Labette | 33S-18E | Marmaton | 28 |
| Labette | 34S-20E | Marmaton | 28 |
| Leavenworth | 9S, 11S-21E | Oread | 16 |
| Leavenworth | 12S-22E | Plattsburg | 19 |
| Leavenworth | 12S-22E | Wyandotte | 20 |
| Linn | 19S-21E | Plattsburg | 19 |
| Linn | 20S-21E | Iola | 22 |
| Linn | 19S-21E | Drum | 23 |
| Linn | 19S, 20S-23E | Dennis | 25 |
| Linn | 20S, 21S-22E | Dennis | 25 |
| Linn | 19S, 21S-24E | Swope | 26 |
| Linn | 19S-25E | Swope | 26 |
| Linn | 22S-23E | Swope | 26 |
| Linn | 21S-23E, 24E | Hertha | 27 |
| Linn | 22S-23E | Hertha | 27 |
| Linn | 23S-23E | Marmaton | 28 |
| Lyon | 16S-10E | Funston | 7 |
| Lyon | 16S-10E | Crouse | 8 |
| Lyon | 16S-11E | Beattie | 9 |
| Lyon | 18S-10E | Beattie | 9 |
| Lyon | 15S, 16S-11E | Grenola and Red Eagle | 10 |
| Lyon | 20S-10E | Foraker | 11 |
| Lyon | 18S-11E | Admire | 11 |
| Lyon | 17S-12E | Wabaunsee | 12 |
| Lyon | 19S-11E, 12E, 13E | Wabaunsee | 12 |
| Lyon | 21S-11E | Wabaunsee | 12 |
| Marion | 20S-4E | Winfield | 2 |
| Marion | 21S-5E | Barneston | 4 |
| Marshall | 2S-7E | Winfield | 2 |
| Marshall | 3S, 5S-6E | Barneston | 4 |
| Marshall | 2S, 5S-9E | Beattie | 9 |
| Miami | 15S, 16S-21E | Stanton | 18 |
| Miami | 16S-24E | Wyandotte | 20 |
| Miami | 17S-22E | Wyandotte | 20 |
| Miami | 17S-22E | Lane | 21 |
| Miami | 17S-22E, 23E | Iola | 22 |
| Miami | 18S-21E | Iola | 22 |
| Miami | 18S-25E | Cherryvale | 24 |
| Montgomery | 32S-14E, 15E | Stanton | 18 |
| Montgomery | 31S-15E | Plattsburg | 19 |
| Montgomery | 33S-16E | Drum | 23 |
| Montgomery | 33S-16E | Cherryvale | 24 |
| Morris | 14S, 16S-7E | Barneston | 4 |
| Morris | 17S-6E | Barneston | 4 |
| Morris | 15S-8E | Wreford | 6 |
| Morris | 16S, 17S-9E | Beattie | 9 |
| Nemaha | 3S-14E | Beattie | 9 |
| Nemaha | 5S-11E | Wabaunsee | 12 |
| Nemaha | 1S-12E | Wabaunsee | 12 |
| Neosho | 27S-18E | Iola | 22 |
| Neosho | 29S-17E | Iola | 22 |
| Neosho | 30S-18E | Drum | 23 |
| Neosho | 27S, 29S-19E | Dennis | 25 |
| Neosho | 30S-19E | Swope | 26 |
| Neosho | 29S-29E | Hertha | 27 |
| Neosho | 29S-21E | Marmaton | 28 |
| Osage | 15S, 16S-13E | Wabaunsee | 12 |
| Osage | 15S-15E | Topeka | 13 |
| Osage | 18S-14E | Topeka | 13 |
| Osage | 14S, 15S-17E | Deer Creek | 14 |
| Osage | 18S-16E | Oread | 16 |
| Pottawatomie | 7S-12E | Wreford | 6 |
| Riley | 7S-6E | Doyle | 3 |
| Riley | 9S-4E | Doyle | 3 |
| Riley | 7S-6E | Matfield | 5 |
| Riley | 8S-6E | Wreford | 6 |
| Riley | 11S-8E | Funston | 7 |
| Riley | 11S-8E | Crouse | 8 |
| Riley | 8S-7E | Beattie | 9 |
| Riley | 10S-7E | Grenola | 10 |
| Riley | 10S-8E | Foraker | 11 |
| Riley | 10S-7E | Wabaunsee | 12 |
| Shawnee | 10S-15E | Wabaunsee | 12 |
| Shawnee | 11S-14E, 15E | Wabaunsee | 12 |
| Shawnee | 11S-14E, 16E | Topeka | 13 |
| Shawnee | 13S-16E | Topeka | 13 |
| Shawnee | 11S, 13S-16E | Deer Creek | 14 |
| Shawnee | 12S-17E | Deer Creek | 14 |
| Wabaunsee | 14S-9E | Barneston | 4 |
| Wabaunsee | 13S-9E | Wreford | 6 |
| Wabaunsee | 15S-10E | Wreford | 6 |
| Wabaunsee | 13S-10E | Beattie | 9 |
| Wabaunsee | 14S-12E | Grenola | 10 |
| Wilson | 27S-13E | Oread | 16 |
| Wilson | 27S, 28S-14E | Douglas | 17 |
| Wilson | 29S-15E | Stanton | 18 |
| Wilson | 30S-14E | Stanton | 18 |
| Wilson | 29S, 30S-16E | Plattsburg | 19 |
| Wilson | 29S-17E | Iola | 22 |
| Wilson | 30S-16E | Drum | 23 |
| Woodson | 25S-13E | Oread | 16 |
| Woodson | 25S-13E | Douglas | 17 |
| Woodson | 25S, 26S-15E | Douglas | 17 |
| Woodson | 25S-17E | Stanton | 18 |
| Woodson | 26S-16E | Stanton | 18 |
| Wyandotte | 10S-23E | Plattsburg | 19 |
| Wyandotte | 11S-23E | Plattsburg | 19 |
| Wyandotte | 11S-23E, 25E | Wyandotte | 20 |
| Wyandotte | 11S-24E | Cherryvale | 24 |
| Wyandotte | 11S-24E | Dennis | 25 |
| Wyandotte | 11S-24E | Swope | 26 |
Kansas Geological Survey, Chemical Composition of Eastern Kansas Limestones
Placed on web Aug. 14, 2009; originally published in June 1956.
Comments to webadmin@kgs.ku.edu
The URL for this page is http://www.kgs.ku.edu/Publications/Bulletins/119_3/index.html