Kansas Geological Survey, Bulletin 86, Part 2, originally published in 1950
Originally published in 1950 as Kansas Geological Survey Bulletin 86, Part 2. This is, in general, the original text as published. The information has not been updated.
The presence of minute quantities of soluble vanadium compounds in structural clay products made from some high-quality Kansas clays often causes a green or yellow discoloration which detracts from the sales value of the products. The Geochemistry Division of the State Geological Survey of Kansas is investigating methods of eliminating this objectionable discoloration. Colorimetric methods are inadequate for determination of the very small amounts of vanadium present in the clay and must be replaced by more accurate and precise methods. A spectrographic method of analysis which provides the needed accuracy and precision has been developed in the Survey laboratories and is described in this paper. Typical analyses of vanadium in several selected Kansas clays are given.
Kansas clays used in the manufacture of structural products, such as brick and tile, are found generally to contain minute quantities of vanadium compounds which become at least partially soluble in water as a result of the firing. The water-soluble vanadium compounds may give rise to an efflorescence which appears as an unsightly green or yellow discoloration on the surface of the clay product when it is exposed to weathering. The discoloration reduces the sales value of the clay products; accordingly, the problem of reducing or eliminating the discolorations is important to the Kansas clay industry. Solution of this problem must be based on knowledge of the quantity of vanadium present in the raw clays and on investigation of what happens to the vanadium in clay under various conditions of firing. Under some conditions discoloration of clay products may occur even if the vanadium content of the clay is as low as 0.001 per cent V2O5 (Plummer and Romary, 1947, p. 28); on the other hand many clays containing considerably larger amounts of vanadium do not effloresce. A needed first step in research on the control of vanadium-produced efflorescence is the finding of a satisfactorily precise means of measuring minute quantities of vanadium compounds. If such technique for accurate analysis can be developed, it may be used both for testing raw clays and clay products which have been subjected to various conditions of firing. The purpose of this paper is to describe methods of analyzing the vanadium content of clays.
The Kansas clays used in this study (Table 1) were selected by Norman Plummer for analysis of their vanadium content, because previous ceramic tests had shown occurrence of a vanadium efflorescence in products manufactured from them. Four of these clays, now being used for brick making, were selected as standards for comparison with the other clays. The four so-called standard clays are (Table 1) BT-3-3, BT-1-MR, C-51-C, and C-51-6.
Table 1--Kansas clays analyzed for vanadium.
| Clay no. | County | Sec. | Township, South |
Range | |
|---|---|---|---|---|---|
| BT-1-MR | Barton | SW SW | 21 | 18 | 13W |
| BT-3-3 | Barton | SW SW | 21 | 18 | 13W |
| C-44-3 | Cloud | SW | 3 | 8 | 3W |
| C-51-C | Cloud | NW NW | 12 | 6 | 3W |
| C-51-6 | Cloud | NW NW | 12 | 6 | 3W |
| EI-43-C | Ellsworth | N2 NE | 25 | 15 | 10W |
| EI-60-13 | Ellsworth | SE SW | 19 | 15 | 9W |
| EI-61-18 | Ellsworth | NE | 19 | 15 | 9W |
| L-10-01 | Lincoln | W2 NW | 10 | 13 | 7W |
| O-4-16 | Ottawa | NW | 14 | 9 | 5W |
| O-31-5 | Ottawa | NE | 7 | 9 | 1W |
| SE | 6 | 9 | 1W | ||
| S-3-8B | Saline | NE | 30 | 15 | 4W |
| W-7-2 | Washington | NW | 15 | 2 | 3E |
The best method of chemical analysis for vanadium occurring in very small quantities in silicate materials is a colorimetric method. The National Bureau of Standards samples listed in Table 2 were analyzed colorimetrically. In the colorimetric method, the vanadium is first separated from iron, titanium, zirconium, and any other elements present which would cause a color to be developed in an acid solution. The vanadium is then oxidized in acid solution with hydrogen peroxide, causing a reddish-yellow to brownish-red color to be developed, depending upon the amount of vanadium present. This color is compared with the colors of standard solutions containing a known quantity of vanadium in order to complete the analysis. The colorimetric determination of vanadium is subject to large variations in accuracy and precision. These variations are due mainly to (1) loss of vanadium from the sample during separation from other elements, (2) the presence of traces of elements other than vanadium which give colors with hydrogen peroxide, and (3) the fading of color developed in the sample. Also, it is difficult to match the color of the sample precisely against the standard (Hillebrand and Lundell, 1929, p. 362).
Careful colorimetric analysis of materials containing less than 0.1 percent V2O5 commonly vary by as much as 30 to 50 percent of the total V2O5 present. Table 2 indicates variations in V2O5 analyses of several U.S. Bureau of Standards samples, as determined by different analysts.
Table 2--Comparison of vanadium oxide (V2O5) analyses of Bureau of Standards samples*.
| Sample no. |
Percent V2O5 by analyst no. | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Average | |
| 69 | 0.03 | 0.02 | 0.03 | + | + | + | + | + | 0.03 |
| 76 | 0.02 | 0.03 | + | 0.014 | + | + | + | 0.02 | 0.021 |
| 77 | 0.03 | 0.035 | + | 0.029 | + | + | + | 0.035 | 0.032 |
| 78 | 0.04 | 0.07 | + | 0.035 | + | + | + | 0.044 | 0.047 |
| 97 | 0.040 | 0.036 | + | 0.044 | 0.04 | 0.040 | + | + | 0.040 |
| 98 | 0.030 | 0.027 | + | 0.025 | 0.02 | 0.025 | + | + | 0.025 |
| *Compiled from the Certificates of Analysis which accompany each Bureau of Standards sample. +Not determined by this analyst. |
|||||||||
The spectrographic technique used at the State Geological Survey of Kansas is a modification of the internal standard method, in which the element of unknown concentration in the sample is compared against an element of known concentration in the sample, the known element being the internal standard. A small sample of the material to be analyzed is placed in a receptacle machined into one end of a graphite rod, which constitutes one electrode. An electric arc is struck between this electrode and another graphite electrode. The heat from the electric arc causes the sample to be heated to incandescence and volatilized. The spectrograph receives a small portion of the light from the incandescent arc, splits the light into a spectrum, and photographs the spectrum. The spectrum appears on the photographic negative as a series of short, parallel black lines. Each element has its own set of lines which appear on the photograph at entirely different places from the lines of any other element. The amount of light received by the film, which depends on the amount of a given element present in the sample, determines the blackness of the lines of that element. One may measure indirectly the blackness of any single line on the film by passing a beam of light through the film and measuring the difference between the amount of light passing through the clear film and through the line. This measurement is made by using a photoelectric cell.
In the internal standard method, by comparing the blackness of a given line of one element in a sample against the blackness of a given line of another element in the sample, one indirectly determines the ratio of the amounts of each of the elements in the sample. Using a number of standard samples in which each of the elements is known, one may determine the mathematical relation between the ratio of the blackness of the lines and the ratio of the two elements. Then a sample in which one of the elements is unknown may be examined spectrographically, and from the mathematical relation the unknown element may be determined. The mathematical relation may be plotted as a graph, or may be expressed analytically as an equation.
The element chosen to be used as an internal standard must be very similar to the unknown element in its spectrographic qualities for the comparison to be valid. That is, the two elements must volatilize at approximately the same rate, and must require approximately the same voltage across the electric arc for the emission of light. In this study, iron was used as the internal standard, because it satisfies these conditions, and because it is present in sufficient quantity in all the clays for a very accurate chemical determination of iron content.
The internal standard method for spectrographic determinations tends to minimize errors introduced by interfering elements and by uncontrolled changes in the operation of the spectrograph. Since the unknown element and the standard element are very much alike, they are affected in the same manner by variations from sample to sample and by variations of the spectrograph operation. Thus the analysis of identical samples will give identical ratios of the amounts of the two elements in each sample; small variations in readings by the operator and small variations in the composition of the theoretically identical samples should produce the only errors of this method of analysis. This ideal is very nearly achieved by the internal standard method.
Once the spectrographic procedure for analysis of an element has been developed, a large number of samples may be analyzed within a comparatively short period of time. The minimum number of samples analyzed for vanadium on our selected standards was 74. A few analyses varied from the mean values of the analyses of the standards by errors almost as great as the deviations of the colorimetric method. However, the great bulk of data permitted statistical interpretation, which indicated the amount of error one could expect in any given value of V2O5 determined. Analytic rather than graphical calculations were used to avoid the loss of accuracy inherent in the plotting and reading of graphs.
The chemical and mineralogical composition of both the synthetic standard clay (Table 3) and other clays to be analyzed should be very similar in order for them to have similar spectrographic qualities for good spectrographic comparison. The characteristics of spectroscopic excitation of a material as brought forth by the electric are are affected by the composition of the material. The spectroscopic buffer (see Preparation of Clays for Spectrographic Use) helps somewhat, but does not overcome all the differences between two dissimilar clay samples. Selection of the standard clay was based upon the similarity of chemical compositions of the standard clay and the clay to be analyzed for vanadium. Table 3 gives chemical analyses of the standard clays.
Table 3--Chemical analyses of standard clays.
| Constituent | BT-3-3 | BT-1-MR | C-51-C | C-51-6 |
|---|---|---|---|---|
| SiO2 | 72.56 | 76.52 | 76.58 | 76.04 |
| Al2O3 | 17.44 | 13.57 | 13.12 | 14.76 |
| Fe2O31 | 1.21 | 1.75 | 3.49 | 1.53 |
| TiO22 | 0.84 | 1.37 | 0.66 | 0.89 |
| CaO | 0.40 | 0.41 | 0.45 | 0.42 |
| MgO | 0.31 | 0.21 | 0.39 | 0.34 |
| P2O5 | trace | trace | trace | |
| SO3 | trace | trace | trace | trace |
| Ignition loss | 5.68 | 4.73 | 4.85 | 5.08 |
| Undetermined difference3 |
1.56 | 1.44 | 0.46 | 0.94 |
| 1Average of at least three special iron determinations. 2Gravimetric determination, RO2 + R2O5 (Runnels, Utter, and Reed, 1950, pp. 51-53). 3Primarily alkalis. |
||||
Thanks and appreciation are expressed to two members of the staff of the State Geological Survey of Kansas, R. T. Runnels, Geochemistry Laboratory, and Norman Plummer, Industrial Minerals and Ceramics Division. Mr. Runnels supplied the chemical analyses and helped with the preparation of most of the samples. Mr. Plummer provided the clays used in the study, and his knowledge of clays and ceramics was always available.
There are several general steps in our spectrographic analysis for vanadium in clays. They include: preparation of synthetic standards, preparation of the clay sample for spectrographic analysis, the actual analysis, and interpretation of data obtained.
Because no suitable standards were available, a set of synthetic standard clays had to be prepared in order to have a basis of comparison for use in testing other clays. The following paragraphs describe the preparation of the synthetic standards. The general procedure was as follows: (1) removal of some of the vanadium from one of the clays to form a matrix containing less vanadium than did the original clay; (2) grinding the matrix; (3) special chemical analyses of the iron content of the standard clays; (4) qualitative spectrographic analysis of the scum removed from the clay; and (5) addition of known amounts of vanadium (V2O5) to weighed portions of the matrix, forming samples which contain the minimum vanadium in the matrix plus known added amounts of vanadium.
Removal of vanadium--All the clay samples to be used as synthetic standards were pulverized in a pan mill and made into standard test bricks about 1 1/8 by 1 1/8 by 7 inches. A small part of each brick was reserved and the remainder was treated with the A.S.T.M. test for efflorescence of soluble salts in bricks (A. S.T.M., 1946, designation C 67-44, p. 191). The bricks were placed on end in beakers and half submerged in distilled water which carried the soluble salts, including those of vanadium, upward in the ceramic material. Evaporation of the water caused the soluble salts to be deposited as a scum at the upper surfaces of the bricks. After several days of this partial immersion, the bricks were removed from the beakers, and the scummed portions chipped off. Thus, some of the vanadium was removed from the bricks being treated.
Grinding the matrix--Next the test bricks (scummed portions removed) were ground for 4 to 8 hours, first with a porcelain mortar and pestle, then in a porcelain ball mill using porcelain pebbles. The ball mill reduced the maximum particle size until a representative portion of the sample could be washed through a 200-mesh screen; the balance of the sample was then assumed to be sufficiently pulverized and mixed for spectrographic use.
The small parts of the bricks which were reserved before the immersion treatment were pulverized in the same manner for later treatment.
Chemical analysis--The clay from which a part of the vanadium had been removed served as a matrix (containing the minimum vanadium) which was almost identical in all other respects to the original fired clay. Because iron was to be used as the internal standard, especially precise chemical analyses for the iron content of both the original clay and the minimum-vanadium clay were made. No change in the iron content was noted.
Analysis of the scum--Qualitative spectrographic analyses of the scum from the bricks showed the main metallic content of the scum to be vanadium. The remainder of the scum was mostly alkalis and alkaline earths. The only major change of the minimum-vanadium matrix from the original clay was decrease in vanadium content.
Addition of known amounts of vanadium--To weighed portions of the minimum-vanadium clay were added small, known amounts of vanadium oxide. By this means, samples containing either more or less vanadium than the original clay but having identical iron content and identical matrices of all other constituents were prepared. For example, to 100 grams of the minimum-vanadium matrix of clay no. BT-3-3 was added 0.0043 grams of V2O5. The vanadium content of this 100-gram portion of the minimum-vanadium clay was thus increased by a known addition of 0.0043 percent V2O5. To similar portions of the minimum-vanadium matrix of clay BT-3-3 were added sufficient V2O5 to increase the unknown minimum vanadium oxide percentages by known additions of 0.0064, 0.0084, 0.0106, and 0.0128 percent V2O5, respectively. In this way seven clay samples of identical character except for their vanadium content were obtained. These comprised (1) the original clay, (2) the minimum-vanadium clay, and (3) the five portions of minimum-vanadium clay to which different known amounts of vanadium had been added. The other three standard clays (Table 3) were treated in a similar manner.
A spectrographic buffer was added to each of the clays to stabilize the arc current when exciting a sample by volatilizing it in the electric arc, and to aid in carrying the sample into the arc. Experimental spectrographic work showed that powdered graphite was not a satisfactory buffer, because it allowed the arc current to become unstable. A more satisfactory buffer was potassium bisulphate, because it kept the are current stable until a few seconds before the sample was completely volatilized. Also, the use of potassium bisulphate prevented the burning of much carbon in the arc, permitting a long period of excitation with no interference from cyanogen bands in the spectra. Therefore, equal portions by weight of clay and potassium bisulphate were mixed, then sintered over a low flame in small porcelain crucibles. The sinter cakes were pulverized with a mullite mortar and pestle, and the powdered samples stored in small glass vials for use in the tests.
The spectrographic equipment used was manufactured by Applied Research Laboratories, and includes:
In addition, a magnetic arc rotator was built and mounted on the optical bench of the spectrograph. The magnetic rotation of the arc causes smoother burning of the sample, which stabilizes the arc current and makes the analyses much more reproducible (Myers and Brunstetter, 1947).
The conditions of sample excitation and the optical settings were adjusted for the different clay samples to provide optimum control of sample volatilization and of quantitative measurements. The average conditions and settings were as follows: (1) exposure time, 60 seconds; (2) light transmission to grating decreased to 0.10 by the rotating sector opening and to 0.383 of the 0.10 by the grating doors, so that the amount of light passed to the grating was very nearly 0.0383 of the maximum available; (3) arc gap, 5.5 mm; (4) current, 11.5 amperes; (5) slit, 50 microns. The average variation of the arc current was about ±0.1 ampere, with variation of ±0.5 ampere during the last 5 or 10 seconds. Because the arc current had to be controlled manually, the arc gap could not be held constant. However, experiments showed that very little of the electrodes burned away until the last 5 or 10 seconds of excitation. During the remaining time, while the last small fraction of the sample volatilized, the gap distance of the electrodes increased 0.5 to 1.0 mm. The lens was set on the optical bench to focus the arc on the grating (parallel rays of light from the lens to the slit for uniform slit illumination). Moving film spectra showed that very little of the iron or vanadium was left to burn during the last 5 to 10 seconds.
Eastman spectrum analysis No. 1 film was used. After exposure, the film was developed for 3 minutes in Eastman D-19 developer, shortstopped 10 seconds in 3 percent acetic acid, fixed 1 minute in Eastman rapid fixer (X-ray), washed 1 minute in rapidly flowing tap water, rinsed 30 seconds in distilled water, and dried for 1 1/2 minutes with an Applied Research Laboratories infra-red film dryer.
Multiple exposures were used to aid in smoothing out the data. By superimposing on the film five exposures of the spectrum of a single sample, one may cause the film itself to reduce the overall experimental error. This required reduction of the rotating sector opening to one-fifth the opening required for a single exposure. The film received the same total amount of light as for a single exposure, but actually recorded the average of five exposures (Fassel and Wilhelm, 1947, p. 1).
Platform electrodes with center posts were made from regular grade spectroscopic carbons; the platform shape provides for a rapid temperature rise to the boiling point of the sample and for less heat conduction away from the sample. This shape also aids in preventing the arc from creeping up the side of the upper electrode. To prevent mechanical loss of part of the sample during handling, a drop of alcohol saturated with sugar was used to cement the sample to the electrode.
The film was calibrated graphically in the region between 3150 and 3250 Angstrom units, as described by Brode (1943, pp. 110-113). Several densitometer readings of background and line transmission values were obtained for each exposure, to ensure that the average readings would be as nearly correct as possible. Background corrections were made from the film emulsion calibration curve (Brode, 1943, pp. 113-114). The lines experimentally selected as best suited for the densitometric readings needed for the internal standard method were V 3183.9 and Fe 3225.79, iron being the internal standard. There was no discernible interference of the vanadium line; the iron line sometimes had interference from a calcium line, Ca 3225.9. The interference was minimized by the use of a spectrograph slit of 50 microns, somewhat less than that recommended by Applied Research Laboratories, but still suitable if the densitometer slit is narrow enough to cover the width of the photographed lines. The ratio of the densitometer galvanometer deflection for the iron line to the deflection for the vanadium line was computed logarithmically from the film emulsion calibration curve (Brode, 1943, pp. 106-110).
The largest source of error in this work was due to a combination of varying sample sizes, varying arc current, variations in film processing, and several other spectroscopic factors. Because these errors are seen only in the measurements of the opacity or blackness of photographed spectra, they are logarithmic in nature. Therefore, any statistical treatment of the data had to be performed upon the logarithms of the deflection ratios, log (D(Fe) / D(V)). The statistical formulae used gave the geometric mean (algebraic mean of the logarithms) of the deflection ratios, and the probable error of the logarithm of the mean deflection ratio. The probable errors were converted to percentages of the mean deflection ratios, making the positive probable error larger than the negative probable error in most cases. The formulae are as follows (Croxton and Cowden, 1939, pp. 221-226, 240-245, 691-705):
(1) Geometric Mean
log G = (log X1 + log X2 + + log XN) / N
where G = geometric mean.
X1, X2, ... XN = deflection ratios of each trial,
N = number of trials.
(2) Deviation from the Geometric Mean
Xi = log Xii - log G, i = 1 to N
where Xi is the logarithmic deviation of the i-th term from log G.
(3) Probable Error (logarithmic)
Because multiple exposures were used, with five superimposed exposures per spectrum, each spectrum must be considered equivalent to five identical items in the statistical distribution. The formula for the probable error becomes
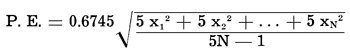
It may be shown that the probable errors of the determined amounts of V2O5 are very nearly equal to the probable errors of their deflection ratios; they will be used as though equal.
In the graphic method of quantitative spectrographic analysis, one plots data for known samples to obtain a working curve. It may be shown from a consideration of the physical phenomena of spectroscopy and densitometry that the analytic relation corresponding to the graphic method is
log (U/S) = a log [D (S) / D (U)] + b,
where U is the concentration of the element to be determined, S is the concentration of the standard element, D is the densitometer-galvanometer deflection of a spectrum line, and a and b are constants to be evaluated.
The constants a and b may ordinarily be determined from spectrographic data on two different standard samples for which S and U are known. In this study, U, the total vanadium concentration, was not known in any of the samples standardized. Hence, at least three samples were required to find the three unknowns, a, b, and U. The three simultaneous equations obtained by getting spectrographic data on the three samples are transcendental equations, involving ordinary numbers and logarithms, and are not subject to algebraic solution. A graphic method was used to obtain an approximation to the solution, and Newton's method for the solution of transcendental equations was used for the final solution.
When a and b have been determined for a set of synthetic standards (Table 4), the V2O5 content of the original sample may be calculated using the equation above. Samples other than the standard samples are analyzed (Table 5) for V2O5 by use of the equation, setting in values of a and b for that standard sample which is chemically most similar to the unknown sample.
Table 4--Analyses of synthetic standards for vanadium oxide (V2O5)
| Clay | No. of samples examined |
Deflection ratio, geometric mean |
Percent V2O5 |
Probable error as percentage of V2O5 |
|
|---|---|---|---|---|---|
| (+) | (-) | ||||
| BT-3-3 (Constants: a = .4327, b = -1.4477) | |||||
| Original clay | 14 | 0.913 | 0.0415 | 5.5 | 5.0 |
| Min. V2O5, x percent |
15 | 0.831 | 0.0399 | 8.8 | 8.2 |
| x + 0.0043% | 15 | 1.004 | 0.0441 | 12.4 | 11.0 |
| x + 0.0064% | 15 | 1.160 | 0.0461 | 9.8 | 8.9 |
| x + 0.0084% | 14 | 1.281 | 0.0483 | 11.1 | 10.1 |
| x + 0.0106% | 14 | 1.432 | 0.0505 | 11.2 | 10.1 |
| x + 0.0128% | 15 | 1.589 | 0.0527 | 8.2 | 7.6 |
| BT-1-MR (Constants: a = .4327, b = -1.4477) | |||||
| Original clay | 20 | 0.528 | 0.0473 | 7.5 | 7.0 |
| Min. V2O5, x percent |
20 | 0.429 | 0.0433 | 9.0 | 8.2 |
| x + 0.0021% | 20 | 0.481 | 0.0455 | 6.8 | 6.4 |
| x + 0.0085% | 20 | 0.699 | 0.0534 | 1.5 | 1.4 |
| x + 0.0171% | 20 | 0.896 | 0.0595 | 1.5 | 1.5 |
| C-51-C (Constants: a = .3331, b = -1.4855) | |||||
| Original clay | 15 | 0.366 | 0.0816 | 9.7 | 8.9 |
| Min. V2O5, x percent |
15 | 0.272 | 0.0740 | 10.5 | 9.5 |
| x + 0.0043% | 15 | 0.312 | 0.0774 | 3.9 | 3.8 |
| x + 0.0085% | 14 | 0.390 | 0.0834 | 1.2 | 1.2 |
| x + 0.0170% | 15 | 0.507 | 0.0910 | 1.4 | 1.4 |
| C-51-6 (Constants: a = .4078, b = -1.4647) | |||||
| Original clay | 15 | 0.896 | 0.0502 | 3.9 | 3.7 |
| Min. V2O5, x percent |
15 | 0.576 | 0.0419 | 10.6 | 9.6 |
| x + 0.0043% | 15 | 0.769 | 0.0472 | 1.8 | 1.8 |
| x + 0.0085% | 15 | 0.959 | 0.0516 | 2.5 | 2.5 |
| x + 0.0170% | 15 | 1.328 | 0.0589 | 6.7 | 6.3 |
Table 5--Analyses of some selected Kansas clays for vanadium oxide (V2O5)
| Clay no. | Percent V2O5 |
Clay used for comparison |
|---|---|---|
| BT-3-3 | 0.0415 | * |
| BT-1-MR | 0.0473 | * |
| C-51-C | 0.0816 | * |
| C-51-6 | 0.0502 | * |
| C-44-3 | 0.144 | C-51-6 |
| EI-43-C | 0.044 | BT-3-3 |
| EI-60-13 | 0.022 | BT-3-3 |
| EI-61-18 | 0.038 | BT-3-3 |
| L-10-01 | 0.037 | BT-3-3 |
| O-4-16 | 0.046 | BT-3-3 |
| O-31-5 | 0.024 | BT-3-3 |
| S-3-8B | 0.064 | BT-3-3 |
| W-7-2 | 0.029 | C-51-6 |
| *Original clays of the synthetic standards, Table 4. | ||
The spectrographic analyses of the clays studied show that all contain vanadium in the same order of magnitude. This indicates that, in general, the spectrographic method for the determination of small amounts of vanadium in clays is valuable for determining which Kansas clays contain troublesome amounts of vanadium. The method can be used along with the many ceramic tests necessary for the evaluation of ceramic raw materials. The probable errors of the V2O5 analyses of the clays, other than the standards, may be assumed to be of the order of ±10 percent, somewhat greater than the probable errors of the standard clays. A clay which has been shown, by comparison with a standard clay, to contain sufficient vanadium to cause possible efflorescence may be analyzed by the method used for the standard clays, if a more accurate analysis is desired. The comparison method will be adequate for most analyses.
Figure 1--Working curve for clays of type BT-3-3 and BT-1-MR.
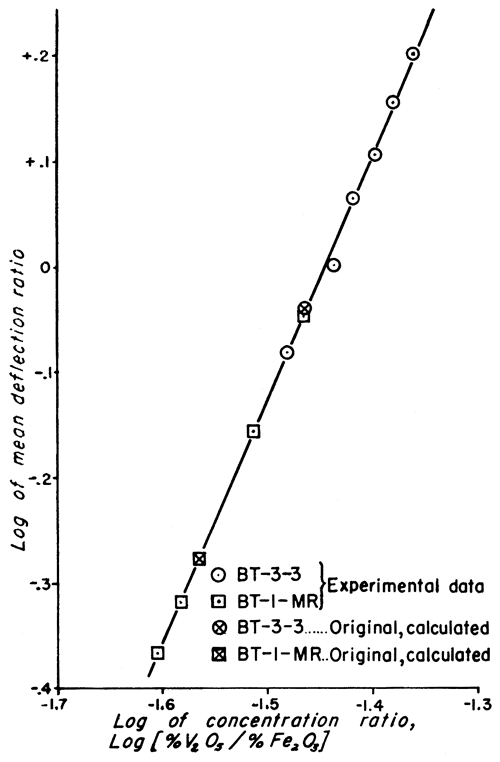
Figure 2--Working curve for clays of type C-51-6.
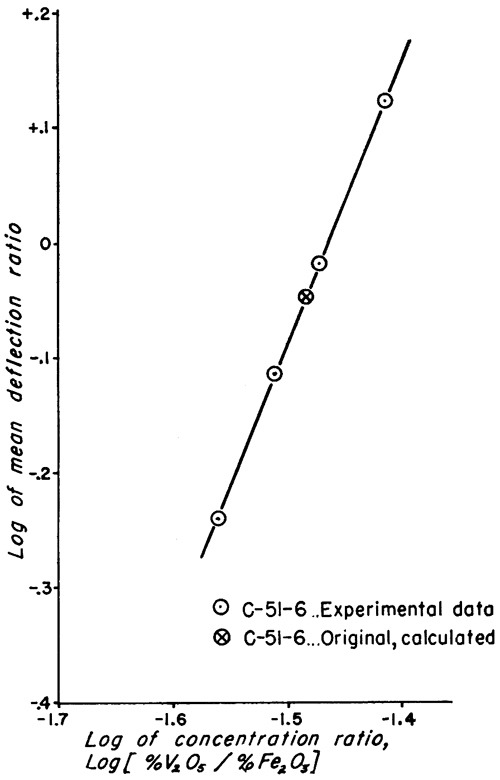
The analytic method used to obtain the vanadium concentration of the standard samples is not as accurate as the method of least squares, used for evaluating physical data; but the discrepancy is very small (Croxton and Cowden, 1939, p. 694). This is especially true for cases in which the plotted data give a straight line (Figs. 1 and 2) because it is relatively simple to estimate the best straight line to draw through the points.
Doubt may be expressed as to whether the precision of the work justifies carrying three significant figures in the vanadium values of the standard samples (Table 4). If the precision were less than the calculated probable error indicates, the mean values of the deflection ratios would more likely be inaccurate. This would cause a much greater scattering of points about the straight lines of the examples. Because the plotted points fall so nearly on the lines, it is very unlikely that the vanadium values listed for the standards are much in error. Considering the number of samples analyzed and the statistical treatment of the data, this was to be expected, assuming the validity of the spectrographic and mathematical methods. Three significant figures were retained for the standard clays, but the probable errors were also listed in order to indicate the trustworthiness of any single value for vanadium.
The probable errors of our V2O5 analyses of the standard clays indicate that a high degree of precision may be obtained, much better than the precision of the usual colorimetric methods. A more suitable spectrographic buffer than the potassium bisulphate is being checked; if it proves satisfactory, the probable error should be reduced further.
The spectrographic method developed for this study (i.e., making synthetic standards and analytic calculations), should be applicable to analysis for other elements difficult to determine by chemical methods when present in small quantities. Also, the method need not be confined to the examination of clays; with suitable sample preparation and spectrographic buffers, most industrial minerals should be adaptable to this method of analyzing for their trace elements.
American Society for Testing Materials (1946) A.S.T.M. Standards, part II, Nonmetallic materials-constructional, pp. 1-1762, Philadelphia, Pa.
Brode, W. R. (1943) Chemical spectroscopy: 2d ed., John Wiley & Sons, Inc., New York, pp. 1-677.
Croxton, F. E., and Cowden, D. J. (1939) Applied General Statistics: Prentice Hall, Inc., New York, pp. 1-957.
Fassell, V. A., and Wilhelm, H. A. (1947) The quantitative spectrographic analysis of the rare earth elements: Iowa State College, Tech. Infor. Div., Oak Ridge Operations, Oak Ridge, Tenn., Publ. no. MDDC-1512, pp. 1-16.
Myers, A. T., and Brunstetter, B. C. (1947) Magnetic rotation of the direct current arc in spectrographic analysis: Analytical Chemistry, vol. 19, no. 1, p. 71.
Plummer, Norman, and Romary, J. F. (1947) Kansas clay, Dakota formation: Kansas Geol. Survey, Bull. 67, pp. 1-241, figs. 1-17, pls. 1-7.
Runnels, R. T., Utter, M. G., and Reed, A. C. (1950) Determination of ferric oxide and titania in presence of alumina: Am. Ceramic Soc. Jour., vol. 33, no. 2, pp. 51-53.
Kansas Geological Survey, Spectrographic Analysis for Vanadium in Kansas Clays
Placed on web Aug. 12, 2009; originally published in May 1950.
Comments to webadmin@kgs.ku.edu
The URL for this page is http://www.kgs.ku.edu/Publications/Bulletins/86_2/index.html