Kansas Geological Survey, Bulletin 191, pt. 1, originally published in 1968

Originally published in 1968 as part of "Short Papers on Research in 1967," Kansas Geological Survey Bulletin 191, part 1, p. 3-7. This is, in general, the original text as published. The information has not been updated.
In studies of water quality, knowledge of trace element composition is important. Cobalt, iron, lithium, manganese, nickel, strontium, zinc, and silicon were determined in water from several streams and ponds of the Kansas River basin. The method has proven to be routinely usable in the range of 0.01-1 ppm for Co, Ni, and Li, 0.01-10 ppm for Mn, Sr, Zn, and Si, and 0.01-100 ppm for Ca, Mg, and Fe. These trace elements and others can be determined from the same concentrated solution by evaporating 500 mL of filtered water down to 10 mL. This solution is then brought up to 50 mL by the addition of distilled, deionized water. The coefficient of variation for each element was Co 6% at 0.08 ppm, Fe 5% at 0.06 ppm, Li 10% at 0.01, Mn 2.5% at 0.02, and Sr 7% at 0.08 ppm levels, respectively. While it is recognized that these coefficients of variation values can be improved, they were deemed adequate for this study.
A study of the trace element content of the rivers in the lower Kansas River basin was undertaken as part of a much larger program directed toward developing a programmed water quality model (O'Brien et al., 1967) for the main stem Kansas River. (Financial assistance for part of this work was provided by a Federal Water Pollution Control Administration grant to one of us [W. J. O'Brien with E. E. Angino] under contract PH 86-66-63 to the Center for Research in Engineering Science, The University of Kansas. T. C. Waugh aided in the development of the method for trace element analysis, and R. L. Kennedy, R. L. Madl, and Mrs. Khawla Sabih prepared the samples.) The purpose in doing the trace element analyses was two-fold: 1) To develop methods for trace elemental analyses, and 2) To collect data on the trace element content of the different rivers in the Kansas River basin. Because little information on trace elements exists for the rivers of this area, it was thought that such an investigation would be worthwhile.
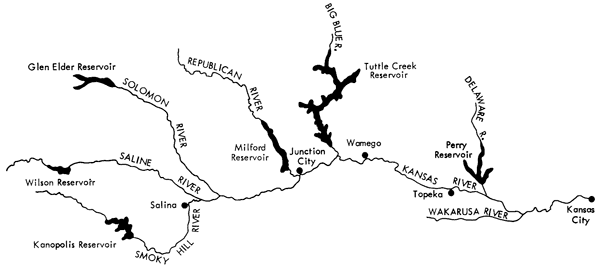
The rivers of the basin that were sampled for this study (Fig. 1) include the Smoky Hill, Saline, and Solomon rivers in the western part of the area, the Republican and Blue rivers in the central part, and the Delaware and Wakarusa rivers in the eastern part. The Kansas River, which extends from Junction City to Kansas City, was also sampled at intervals of 20 to 40 miles.
Figure 1--Map of the portion of the Kansas River basin from which samples were collected.
All samples analyzed for trace elements were prepared in the following manner. Upon being received in the laboratory the water was filtered through a 1.2μ Millipore filter to remove any suspended material. Five hundred mL of the filtered water (to which was added 10 mL of hydrochloric acid) was then evaporated down to a volume of approximately 10 mL, transferred to a 50 mL volumetric flask, and diluted to the mark with distilled, deionized water. This gave a solution with a concentration factor of 10. The acid was added to keep the SiO2 and iron in solution. Diluting the concentrated water to 50 mL with methanol was also tried. According to Wheat (1964), the use of an 80% water methanol solution will increase the sensitivity three to four times over straight aqueous solutions. This can be helpful where low concentrations of a particular element are present. However, a drawback exists to the use of methanol solutions. It was found that when methanol was used as a diluent, fresh standards had to be made up for each series of analyses. With aqueous solutions, standards, which ranged from 0.1 to 10 ppm, could be kept an average of 5-8 weeks before any change was noted. Iron was an exception to this average. Solutions of 100 ppm or less could not be kept for more than one day. For the bulk of the samples run during this study, the methanol dilution step was not necessary. Adequate sensitivity was obtained using the aqueous solutions. The elements routinely determined on the concentrated water solutions were Co, Fe, Li, Mn, Ni, Si (as SiO2), Sr, and Zn. In addition to these elements, Ca and Mg were determined separately. Due to the high concentration of Ca and Mg, it was necessary to dilute the original water sample 25 times in order to complete a successful instrumental analysis.
The instrument used for the determinations was a Jarrell-Ash model 82-500 series atomic absorption spectrometer. The instrument utilizes a 0.5-m Ebert monochromater and, for our determinations, was equipped with a 100-μ entrance and a 150-μ exit slit. The burner used was a Jarrell-Ash triflame burner, which consists of a laminar flow head mounted over a Hetco burner. A fuel combination of air-H2 was used for all determinations except SiO2. A fuel combination of acetylene-nitrous oxide was necessary to determine SiO2 successfully. Fuel and support gas pressures were adjusted for maximum sensitivity for each element.
Twelve locations within the Kansas River basin were sampled once every month from June 1966 through May 1967. The elements studied, the concentration ranges measured, and the calculated coefficient of variation for each element are summarized briefly in Table 1.
Table 1--Concentration ranges and calculated coefficients of variation of the elements studied.
| Element | Concentration range, ppm |
Coefficient* of variation |
|---|---|---|
| Ca | 10.0-176.0 | |
| Mg | 8.0-54.0 | |
| Sr | 0.012 -0.3 | 7.0% at 0.08 ppm level |
| Co | 0.0-0.3 | 6.00%, at 0.08 ppm level |
| Ni | 0.003-0.12 | |
| Fe | 0.013-0.8 | 5.00% at 0.06 ppm level |
| Mn | 0.003-0.7 | 2.5 0% at 0.02 ppm level |
| Zn | 0.01-0.5 | 0.350% at 0.40 ppm level |
| Li | 0.0-0.04 | 10.0% at 0.01 ppm level |
| SiO2 | 5.9-31.3 | 2.3 0% at 7.0 ppm level |
| * Coefficient of variation was calculated on the basis of 12 determinations. | ||
The primary interference encountered in the determination of calcium and magnesium came from sulfate (SO4). The sulfate content of the river waters was as high as several hundred ppm at some locations. This interference was eliminated with the addition of 1500 ppm strontium as SrCl2. There were no interferences encountered during this study that were not eliminated by the addition of strontium.
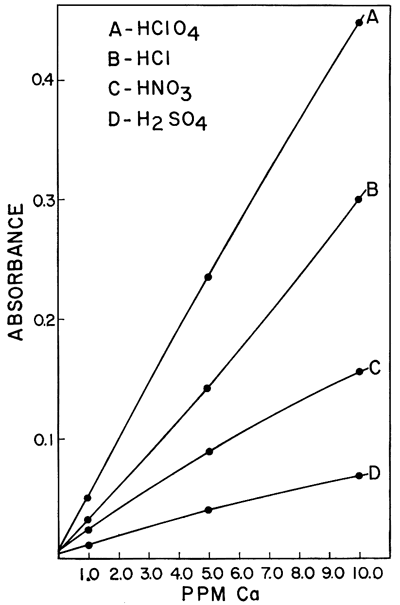
One of the interferences which was found to be of importance was the effect of different acids upon the sensitivity of calcium and magnesium in a hydrogen-air flame. Galle and Angino (1967) reported a difference in standard curves obtained when four different acids were used to put the calcium and magnesium into solution. The acids studied were HClO4, HCl, HNO3, and H2SO4. The work was done with a Hetco (total consumption) burner and a hydrogen-air fuel mixture. Figures 2 and 3 show the results obtained for calcium and magnesium, respectively. It was felt that this effect was probably caused, in part, by the type of burner in the system. It was also felt that the use of a laminar flow type burner would eliminate much of the differences demonstrated by these two figures. However, this is not true if a low-temperature hydrogen-air flame is used. Figure 4 shows the results obtained using the same concentrations of calcium as shown in Figure 2 but doing the determinations on a laminar flow type burner and using a hydrogen flame. As can be seen, the differences still remain, but the net effect was to increase the sensitivity in each case. As is commonly known, the differences can be eliminated by use of a hotter flame (e.g., an acetylene-air flame). This eliminates the difference between HClO4, HCl, and HNO3. However, a difference in the H2SO4 curve remains, due, most likely, to the sulfate interference. The hydrogen-air flame was used when this work was begun, and its use continued because it produces less recorder noise than an acetylene-air flame. The sensitivities obtained for calcium and magnesium are adequate for our needs and as long as standard and sample solutions are made up with the same acid, no viscosity or other problems were encountered with the Ca and Mg determinations.
Figure 2--Comparison of the effect of four acids upon the absorption of calcium using a hydrogen-air flame and a Hetco burner.
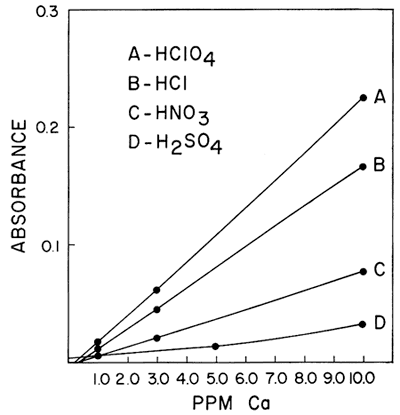
Figure 3--Comparison of the effect of four acids upon the absorption of magnesium using a hydrogen-air flame and a Hetco buriier.
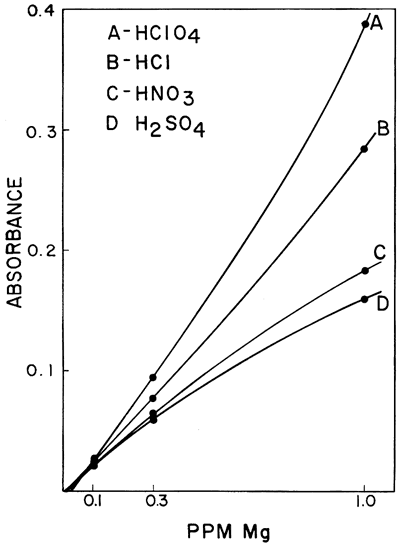
Figure 4--Comparison of the effect of four acids upon the absorption of calcium using a hydrogen-air flame and a laminar flow burner.
The concentrations of the trace elements, as determined for this study, appear to have seasonal variations. The highest concentrations of Co, Fe, and Ni were found during the months of July, August, and September. The concentrations of these elements, in general, decreased as the weather got colder. The concentrations of Li, Mn, and Sr, however, showed a tendency to increase during the winter months of December, January, and February. The data for Zn and SiO2 are incomplete and go back only to December of 1966; therefore, no information about seasonal variations is available for them.
A technique used for SiO2 determinations might be of interest. The cathode power supply of the Jarrell-Ash atomic absorption units may be operated in two different modes. In the normal mode of operation a dc current drives the cathode tube, which is used in conjunction with a mechanical chopper to provide a signal to the monochromater of 87 cycles per second. The second mode of operation involves changing the input current to the cathode tube from dc to pulsed dc. This is done on the Jarrell-Ash instrument by simply moving a switch from the "normal" position to what is called a "high intensity" position. In this position the signal is pulsed electronically at 87 cps. This in effect provides a signal of high intensity for short intervals maintaining a mean current below that of the normal hollow cathode rate. High intensity provides an output 5-20 times as intense as the mechanically pulsed dc output. It was found that by using the high intensity mode of operation for the SiO2 determinations, most of the signal noise in the recorder which was present when using a scale expansion of 8-10 mv was eliminated. When using the high intensity mode, the sensitivity is reduced; but when most of the determinations involve SiO2 in concentrations of 10 ppm or greater, the reduction in sensitivity does not appear to be critical. This method was tried in all determinations in which the recorder scale expansion was needed and an excessive amount of signal noise was present (particularly in determinations of Co and Sr). The technique was successful in reducing the noise level so that readings could be made more accurately.
Another statement on SiO2 determinations by atomic absorption can be made here. Presently, we are working on a method for the determination of SiO2 and Al2O3 in carbonate and silicate rocks. This method involves fusing a powdered sample with lithium metaborate as described by Ingamells (1966). After the sample is fused, it is taken into solution with H2O and nitric acid. The solution technique appears to be successful and our preliminary results on USBS and USGS standard samples are encouraging. The method, however, breaks down with those samples which have 80-100 percent SiO2. In these cases it is difficult to get all of the SiO2 into solution. However the technique appears to work for most other samples, particularly carbonates.
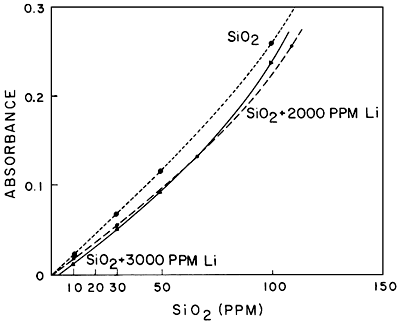
As far as interferences are concerned, little information in the literature exists as a guide. Calcium appears to interfere slightly and lithium in large amounts causes some interference. Figures 5 and 6 demonstrate these interferences. Iron apparently does not interfere, at least at the levels encountered during this investigation. Aluminum may be expected to cause some interference, although this has not been checked to date.
Figure 5--Comparison of standard curves of SiO2 and SiO2 plus 50 ppm Ca.
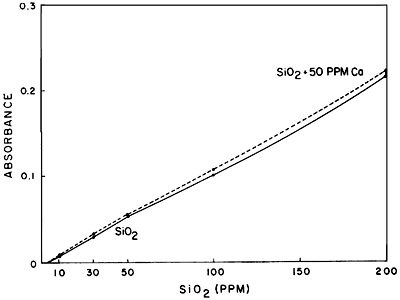
Figure 6--Comparison of standard curves of SiO2 and SiO2 plus 3000 ppm and 2000 ppm Li.
The highest concentration of trace elements is found in the western part of the region, in the Smoky Hill, Saline, and Solomon rivers. The concentration of the trace elements determined, in general, gradually decreases and is the lowest at the eastern end of the region or the lower portion of the Kansas River near Kansas City. Apparently most of the trace elements found in the Kansas River itself are contributed by the Dakota Formation through which the Smoky Hill, Saline, and Solomon rivers flow.
Very little in the way of trace elements is contributed east of the point where the rivers join to form the Kansas River. The fact that the elemental concentrations are diluted by the time they reach the eastern end of the Kansas River could explain why such low concentrations of the elements are found at the eastern end of the region.
Galle, O. K., and Angino, E. E., 1967, Determination of calcium and magnesium in carbonate and silicate rocks; in, Short Papers on Research in 1966, D. E. Zeller, ed.: Kansas Geological Survey, Bulletin 187, pt. 1, p. 9-11. [available online]
Ingamells, C. O., 1966, Absorptiometric methods in rapid silicate analysis: Analytical Chemistry, v. 38, no. 9, p. 1,228-1,234.
O'Brien, W. J., Angino, E. E., Waugh, T. C., and Stoltenberg, G. A., 1967, Water quality in the Kansas River, now and in the future: Transactions of the 17th Annual Conference on Sanitary Engineering 1967, Bulletin of Engineering and Architecture, University of Kansas Publication No. 57.
Wheat, J. A., 1964, Determination of metallic impurities in water by atomic absorption spectrometry: Office of Technical Services, U.S. Department of Commerce, DP-879 [a report by DuPont on contract AT (07-2-1) AEC Research and Development Report TID-4500], 15 p.
Kansas Geological Survey, Short Papers on Research in 1967
Placed on web Aug. 16, 2011; originally published in April 1968.
Comments to webadmin@kgs.ku.edu
The URL for this page is http://www.kgs.ku.edu/Publications/Bulletins/191_1A/index.html