
|
Original published in D.F. Merriam, ed., 1964, Symposium on cyclic sedimentation: Kansas Geological Survey, Bulletin 169, pp. 21-30 | |
Texas A & M University, College Station, Texas
It is striking that a large percentage of all trace element studies of sedimentary units made to date have been directed toward noncyclic deposits. This discussion attempts to focus attention upon the possible presence of trace element cycles, correlative with the normal cyclic sedimentary deposits.
Although the presence of a given trace element, of itself, will probably not be of value, groupings of trace elements such as Cu, Zn, Pb, Mo, V, Ni, and others can furnish valuable information concerning probable physicochemical conditions prevailing in a given environment. The approach used here also emphasizes the dearth of information available concerning the distribution of trace elements within the separate fractions contributing to the make-up of any rock unit.
It is imperative that we obtain considerable more data pertaining to distribution of trace elements in the separate fractions before it is possible to form a complete understanding of any specific paleoecologic study.
The study of cyclic deposits, especially the well displayed Pennsylvanian and Permian cyclothems of the Midcontinent region, has interested geologists for many years. Not the least studied aspect of these deposits has been the search for possible mechanisms to explain their rhythmic occurrence. Moore (1936) and many others since have discussed the various stratigraphic observations that pertain to questions of continental tectonics and other features related to these questions. However, implicit in all discussions concerning the reoccurrence of rocks of different physical types and lithologies (i.e. sandstone, shale, limestone, sandstone, etc.) is the fact that when examining these cyclic deposits we are (as has been emphasized many times) also confronted with reoccurrence of specific paleoecologic or environmental conditions. In the latter situations, the biogenic element, represented now as fossils, and sediment types now present were dependent in many ways on chemistry of both the water overlying the area and the chemical processes occurring within the sediments themselves. It is pertinent to ask if there is not also a periodicity of elemental content present, in addition to the commonly observable physical cycles. The answer is obviously yes, especially if we consider the sequence of limestone, shale, sandstone, limestone, etc.
That the major elements such as calcium in limestones and silicon in sandstones definitely reoccur in a cyclic pattern is so obvious that we seldom think about the matter, but what of the trace elements and organic compounds present at the time of deposition? What trace elements are likely to be preserved? What trace elements are likely to be concentrated under the different environmental circumstances? What is the relation of the once living forms to present trace-element content? Considering our lack of detailed information on both trace-element distribution and the parameters controlling their distribution patterns in recent sediments, these and similar questions are not always easily answered. Goldberg (1957, 1961a, 1961b), and Krauskopf (1955) have treated this subject in great detail. The present brief review and discussion has borrowed frequently from these and other reports. Here, however, I want to focus attention on what possibly could be called "trace-element cycles" and their relation to periodicity or cyclothemic deposition.
In the study of any series of cyclic deposits it is essential that consideration be given not only the stratigraphic aspect of cyclic deposition, but also that of environments represented by these deposits. Trace-element studies tentatively can be of considerable help in elucidating what environmental conditions may have prevailed during deposition.
Acknowledgments--I wish to express my sincere thanks to Louis S. Kornicker, Donald W. Hood, and Sayed Z. EI-Sayed for their helpful suggestions and criticisms.
Time and financial assistance for this review was made possible by ONR Contract Number NONR-2119(O4), Project 286-E of the Texas A & M Research Foundation.
It is rather striking that a large percentage of all trace-element studies of sedimentary units made to date have been directed toward noncyclic deposits. Let us examine therefore some of the information available and at the same time attempt to clarify within the framework of trace elements in cyclic deposits, some of the questions posed.
For our purposes here I have arbitrarily defined a trace element as one present in concentrations of less than 5 ppm in sea water. The data in Table 1, collected from many sources, illustrate the great difference that exists between the amount of these elements supplied to the oceans and actual concentrations (soluble) observed in sea water. Admittedly, the values for trace-element concentrations in sea water will be refined in the future as more sophisticated methods of analyses become available; however, these data are at least representative.
Table 1--Geochemical balance of some elements in sea water.
| Element | Amount supplied to sea water, ppm (total)1 |
Amount present in sea water, ppm (soluble)2 |
Removal factor | Enrichment factor 3 |
|---|---|---|---|---|
| Si | 166,320 | 4 | 41,600 | |
| Al | 48,780 | 0.002 | 24,400,000 | 200,000 |
| Fe | 30,000 | 0.02 | 1,500,000 | 86,000 |
| P | 708 | 0.1 | 7,080 | |
| Mn | 600 | 0.004 | 150,000 | 41,000 |
| F | 540 | 1.4 | 385 | |
| Rb | 186 | 0.2 | 930 | |
| Ba | 150 | 0.05 | 3,000 | |
| V | 90 | 0.0003 | 300,000 | >280,000 |
| Zn | 79 | 0.007 | 14,000 | 32,500 |
| NI | 48 | 0.0013 | 37,000 | 41,000 |
| Ca | 42 | 0.001 | 42,000 | 7,500 |
| Li | 39 | 0.1 | 390 | |
| Sn | 24 | 0.003 | 8,000 | 2,700 |
| Co | 14 | 0.0001 | 140,000 | 21,000 |
| Pb | 10 | 0.0011 | 9,100 | 2,600 |
| Mo | 9 | 0.009 | 1,000 | |
| Ga | 9 | 0.00001 | 900,000 | 800 |
| Bi | 0.12 | 0.0002 | 600 | 1,000 |
1. Data primarily from Rankama and Sahama (1950).
2. Representative reported value. (primarily from Hood, 1963).
3. Enrichment factors of given elements in some marine organisms over sea water values (above data compiled from many sources including Goldberg, 1957).
To emphasize this difference, we have defined a term which we call the removal factor (column 3). This figure, a ratio for a given element of total river input to the concentration of the element reported in solution for sea water, illustrates clearly the almost quantitative removal of these elements from the marine hydrosphere. An approximation of amount of suspended material that may be present in some instances can be determined from Table 2. To a geologist the most significant figures of the table should be these removal factors. Inasmuch as the excess of input concentration over that presently in solution for any of the elements listed is obvious, a reasonable query might be "where does it go, and how does it get there?" Two mechanisms are dominant (1) fixation by shell-bearing organisms, "biogenic fixation," and (2) fixation into and onto sediments and uptake on suspended matter.
Table 2--Order of abundance in percent, Sigsbee Deep samples1 (93° 53' W., 24° 27' N.). Looked for but not detected (semi-quantitative analysis) Bi, Zr, Sb, W, Cd, Li, Nb; analyses by quantitative emission spectroscopy.
| Element | Depth 10m2 |
Element | Depth 600m |
Element | Depth 1000m |
Element | Depth 3400m |
|---|---|---|---|---|---|---|---|
| Fe | 23.60 | Fe | 34.50 | Fe | 40.70 | Fe | 36.50 |
| Ca | 3.90 | Ca | 3.20 | Cu | 2.50 | Ca | 3.20 |
| Mg | 3.50 | Mg | 2.30 | Ca | 1.80 | Mg | 2.30 |
| Na | 3.00 | Na | 1.80 | Mg | 1.00 | Cu | 2.00 |
| Zn | 0.80 | Zn | 0.90 | Zn | 1.00 | Na | 1.80 |
| Al | 0.70 | Al | 0.60 | Al | 0.40 | Zn | 1.10 |
| K | 0.30 | Pb | 0.32 | Sn | 0.40 | Al | 0.40 |
| Cu | 0.20 | K | 0.20 | Na | 0.20 | Sn | 0.27 |
| Sn | 0.20 | Cu | 0.20 | Pb | 0.20 | Pb | 0.30 |
| Ti | 0.10 | Sn | 0.11 | Mn | 0.040 | K | 0.20 |
| Mn | 0.10 | V | 0.10 | Cr | 0.040 | V | 0.090 |
| Pb | 0.10 | Mn | 0.050 | V | 0.070 | Cr | 0.040 |
| V | .0.060 | Cr | 0.020 | K | 0.030 | Mn | 0.030 |
| Cr | 0.040 | Ti | 0.020 | Ti | 0.020 | Ti | 0.020 |
| Sr | 0.015 | Sr | 0.010 | Sr | 0.010 | Sr | 0.010 |
| Ba | 0.010 | Ba | 0.007 | Ba | 0.007 | Ba | 0.007 |
| Ni | 0.008 | Ni | 0.003 | Ni | 0.003 | Ni | 0.004 |
| Mo | <0.001 | Mo | <0.001 | Mo | <0.001 | Mo | <0.001 |
1. As percent of material making up filter cake.
2. m = meters.
The part played in the respective elemental cycles by marine organisms can be seen by referring to column 4 of Table 1, where enrichment factors for some of the elements in some marine animals are presented. It should be understood, however, that the detailed mechanisms of removal of trace elements by organisms and their subsequent fixation into sediments or into the biogenic phase is incompletely known at present.
A detailed discussion of trace-element content of marine organisms can be found in Vinogradov's (1953) monumental and classic work. The chemistry of the environment in which an animal lives may be reflected in some way in the chemical composition of the organisms. In other words, those marine organisms which contain a high concentration of a given trace element relative to another can probably live and develop only in an environment which supplies this element. For example, some oysters appear to flourish in marine waters with a higher than "normal" copper content. An upper limit, however, does exist beyond which the Cu has a deleterious effect. Although the Cu is primarily fixed in the soft parts of the animal, it may upon demise of the animal become fixed onto humic colloids and thereby be incorporated into sediments (Drozdova and Yemelyanova, 1960). As a consequence of the likelihood of such transfers occurring, we must, then, in attempting to ascertain whether the periodicity of certain stratigraphic units is chemical as well as lithologic, take cognizance of the effect of included fauna (fossils).
In discussing the trace-element content of a given rock sample, the biogenic makeup is all to often ignored. "Grind up the sample and analyze it" appears to be the general approach, prior to trace-element analyses. If trace-element data is a valuable tool in reconstructing paleoecologic conditions, and it can be, then consideration must be given to differentiating between the respective trace-element content of the included fossils, organic fraction, and the enclosing matrix material in the different cyclic deposits.
It has long been known that the concentration of certain heavy metals is greater in marine biosphere than hydrosphere. Ascidians, for example, contain up to 6,500 ppm vanadium (Goldberg, 1957) and 75 ppm niobium; both elements are apparently organically bound.
Recently, Nicholls and others (1959) in a study of zooplankton demonstrated the selectivity in enrichment of certain forms in this group. Lead (Pb) is concentrated in copepods, Mo in crustaceans, Co in chaetognaths, Cd in squids, Rb (Vinogradov, 1953) in algae and other aquatic plants. Noddack and Noddack (1939) presented data on heavy metal content (as percent dry matter) for the coelenterates Cyanea capillata and Metridium diathus. They identified the following metals: Ti, V, Cr, Mo, Mn, Fe, Co, Ni, Cu, Ag, Au, Zn, Cd, Ga, Ge, Sn, Pb, As, Sb, and Bi. Nicholls and others (1959) reported high Pb (65-200 ppm) and V (16-85 ppm) in ash in pteropod shells, and more recently Osterberg (1962), utilizing fallout activity of the fission products Zn65, Zr95, Ru103, and Ce141, found concentrations of these elements in Euphasisia pacifica which suggests that "krill" alone may be an important means of concentration for these trace elements. (Note: Further work of this nature has been reported by Fukai and Meinke (1962) who used activation analysis to study concentration of V, As, Mo, Re, W, and Au in marine biological ash from Ulva sp., Porphyra sp., Tapes japonica, Pandalus sp., and Pneumalophorus japonicus.)
It is reasonable to expect that with increasingly sensitive analytical techniques all the stable chemical elements and their isotopes will be identified in living organisms as Vinogradov (1953) suggested.
In any evolving or growing organisms, the incorporation of a particular element in its hard or soft parts is probably a function of "availability" and physiocochemical properties (thermodynamic stability, solubility, electronegativity, etc.) as well as the need of the organisms. Of these, the most likely governing factor is the presence in certain organisms of concentrator mechanisms developed in response to a need for certain elements in the metabolic processes. Although the absolute abundance of many trace elements varies seasonally, laterally and vertically within certain ranges, their abundance can be considered essentially constant. Nonetheless, given favorable conditions, biogenic activity can accomplish remarkable feats of trace-metal concentrations. The enrichment factors listed in Table 1 demonstrate this well.
As Schubert (1954) pointed out, the relative concentration of metals in the marine biosphere as compared to that in sea water essentially parallels the stability of chelation between metal ions and a variety of coordinated compounds.
Coordinated groups or complexes are commonly called ligands (Kleinburg and others, 1960). For a lucid discussion of ligands and the chelation theory consult the treatise by Martell and Calvin (1952) or the recent text by Kleinburg and others (1960).
For divalent ions, Schubert (1954) gives the following general order of increasing stability (independent of the metal attachment to O2, Ni, or S atoms): Pb > Cu > Ni > Co > Zn > Fe > Cd > Mn > Mg > Ca > Sr > Ba > Ra. Interestingly, Bowen and Sutton (1951) report the following metal concentration values for sponges Cu-1400, Ni-420, Co-50, Mg-.07, and Ca-3.5 ppm.
Clearly, then, biogenic forms act as important reservoir agents for uptake and fixation of trace metals, either in particulate, ionic, or complexed form. Inasmuch as most of the shell material and organic debris present in sea water eventually settles to the bottom and becomes incorporated into the sediments, the importance of coordinated compounds in bringing about fixation of many trace elements in the sediments cannot be overestimated.
Recent advances in organic geochemistry have led to identification of many organic compounds in sediments, Porphyrins (Levorsen, 1954), amino acids (Prashowsky and others, 1961; Abelson, 1959), sugars (Degens and others, 1961), furfurrals (Swain, 1961), and others. What is commonly overlooked, however, is the ability of these components to chelate or fix different trace metals and thereby form stable, relatively insoluble metal complexes. Copper complexes (Drozdova and Yemelyanova, 1960), zinc and mettalloenzymes (Lamb and others, 1958), amino acids, peptides, and proteins (Curd and Wilcox, 1956), vitamin B12, a cobalt chelate (Martell and Calvin, 1952), are just a few of the metal complexes which given favorable geologic conditions may either survive intact for geologically long periods of time, or subsequently by their degradation, transfer included trace-metal load to the sediments.
For example, structural studies (Bailer, 1956) of chlorophyll and haemin show that these substances are complexes of porphyrin family. Much of Ni, Y, and Mo, commonly found in trace amounts in crude oils probably owes its presence to formation of stable porphyrin complexes formed by reaction and replacement by these elements of less stable Fe and Mg porphyrin complexes. The geochemical significance of these complexes and their persistence under certain physicochemical conditions is obvious.
For the above reasons a reasonably distinctive trace-element periodicity in all probability might be present and related to specific lithologic cycles. However, before this possibility is elucidated, it is necessary that greater attention be given to a study of various components making up a rock unit. We need separate trace-element analyses of the rock matrix, included fossils, and included organic fraction.
With such analyses we may then, utilizing information supplied by biochemical, bacteriological, and biologic studies on recent marine forms, be able to delineate more distinctly some of the detailed environmental conditions prevailing during deposition of different types of lithic units.
Krauskopf (1956) on the basis of extensive laboratory studies of some factors that might control concentration of AI, Co, Cr, Cu, Mn, Fe, Mo, Ni, Pb, Ti, Y, W, and Zn in sea water showed that sea water is greatly undersaturated with regard to these ions and suggested that some of the elements are selectively removed from the solution by reactions between ions in solution and solid material, both particles (e.g. clays) in suspension and bottom sediments. What is needed, however, is more information pertaining to distribution patterns for these and other trace elements as related to sediment type. Other than the review by Krauskopf (1955), the only detailed studies between Mo, W, Cu, for example, and silt, clay, and sand-sized fractions of bottom sediments are those of Isayeva (1960), Drozdova and Yemelyanova (1960), and EI Wakeel and Riley (1960). Recently Hirst (1962) discussed the distributional pattern of these elements in sediments from the Gulf of Paria. Although some progress is evident in attempts to delineate what parameters control trace-element abundance and distribution in recent sediments, we still have only meager data available as to the oceanic content and the form in which many specific trace elements are present. Osterberg and others (1963) in a recent study, utilizing radioative Cr51, proved presence and uptake of Cr in the silty sediments off the mouth of Columbia River, suggesting thereby that Cr is probably taken up on clay minerals as suggested by Hirst (1962).
That suspended material high in trace-element content also can be identified in deep sea area was shown in recent work by the author and others in the Sigsbee Deep, Gulf of Mexico. Over 6000 gallons of sea water from four separate depths were pumped through 0.45 micron millipore filters, and the filter cake examined quantitatively by emission spectrography for trace-element content (Table 2). Rough calculations indicate that Fe content at 1000m represents approximately 1 ppm. Other constitutents can be related to this figure. This material will eventually settle out and become a part of the sedimentary blanket covering the floor of Sigsbee Deep. The study of the data collected during this investigation is still in progress. These preliminary results are mentioned here to illustrate that in a study of the factors governing presence of a given trace element in a given environment, the effect of suspended, as distinct from soluble, material must not be overlooked.
Temperature, salinity, pH, Eh, ionic potential, pCO2, etc., all have, as is commonly known, some influence and control on any sedimentary environment, past or present. The effects of these parameters on sediments have been discussed many times (Krumbein and Garrels, 1952; Garrels, 1960; Carrol, 1959; Krauskopf, 1955).
However, inasmuch as these same factors control to a great degree the physicochemical conditions prevailing in the water, and indirectly then the availability of both the different trace elements that are essential to life processes of marine organisms and the means of incorporating these same elements into the sediments, we ought to expect that when certain trace elements or groups of elements are identified they will be reflective of specific environmental characteristics.
In the broadest sense, almost any element can be expected to occur in any lithic unit, however, under a given set of lithologic conditions, some are more likely to be present than others. For a detailed discussion of the distribution of trace metals in sedimentary rocks see Krauskopf (1955).
Significantly, the major carbonate minerals, calcite, aragonite, and dolomite, take into solid solution few trace elements. Of these, strontium, manganese, iron, barium, and lead are most likely to be present in significant amounts. It is significant that the largest contribution to the make up of carbonate rocks is of biogenic origin; parts of some limestones (the Cottonwood Limestone (Permian) of Kansas is typical) are composed essentially of remains of a single organism--in this instance fusulinids.
The abundance of trace elements in carbonate rocks is primarily dependent on the noncarbonate fraction, including detrital and authigenic minerals, noncarbonate skeletal material, organic matter, and trace elements adsorbed upon these materials. The relative contribution to measured trace-metal content of the respective fractions present in carbonates is only imperfectly known at present. This area of study promises to be a fruitful one for future investigations.
Most trace element analyses of carbonate rocks reported to date have been of "total sample" type (Ostrom, 1957; Runnels and Schleicher, 1956; Ronov, 1956).
Recently, Gehman (1962) made an extensive study of organic matter in limestones and shales, with interesting results. He showed that ancient shales have approximately four times more organic matter than limestones (Fig. 1). Further, recent lime sediments have a greater organic content than their older equivalents.
Figure 1--Total organic content of ancient limestones and shales (from Gehman, 1962).
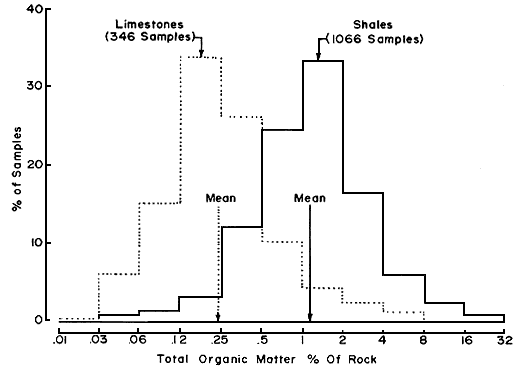
Considering our previous comments relating to trace element fixation by organic means, it is reasonable to ask what percentage of trace-metal contents reported in the literature for any limestone was contributed by its organic fraction.
The uranium content of carbonate sediments is, for example, quite low. What possible environmental conditions might contribute to this? The high HCO3- concentrations and oxidizing conditions prevalent in the depositional areas of carbonates precludes the presence of large concentrations of uranium. Shallow waters high in dissolved oxygen and CO2 will be suitable for formation of organically precipitated material (shells), the major component of many carbonate deposits, but unsuitable for inorganic precipitation of uranium. In this type of environment; the oxidized uranyl ion forms a poorly dissociated tricarbonate complex [UO2(CO3)3]-4 with CO3= or HCO3-. This complexed uranium will then remain in solution and be transported until it reaches a reducing environment or area of lowered pCO2 (the exact opposite of a carbonate environment) where it will precipitate (Bell, 1954). Many other examples could be described, but this illustrates the problem. Within carbonate material itself, most of the uranium is probably tied up in the organic fraction. With exception of analyses of Runnels and Schleicher (1956) which were total rock analyses, little work of this nature has been attempted on the classic cyclothemic deposits of the Midcontinent region.
High uranium content, on the other hand, is normal in marine black shales. Here, reducing conditions preserve much of organic debris that settles to the ocean floor. Uranium is tightly fixed to the humic (rather than sapropelic) component of the carbonaceous material. The humic acids in solution react with uranium to form humates which are precipitated upon lowering of the concentration of divalent cations present (Vine and others, 1958). Such conditions are also favorable for fixation of V, Mo, Ni, Cu, and Zn. Recently Isayeva (1960), Hirst (1962), and others demonstrated a correlation between concentration of these elements and percentage of silt, fine silt, and clay present in a protoshale type of environment.
It has been shown (Vine and others, 1958) that sapropelic matter in marine sediments increases seaward owing to contributions to total organic matter present by remains of marine plankton (largely sapropelic). The most likely environments for deposition and preservation of such organic rich sediments (with large concentrations of humic matter) are nearshore marine, deltaic, and also some nonmarine areas.
Uranium concentrations also show correlation with the abundance gradient of colloidal-sized particles in marine sediments owing to possible presence of U in clay minerals (in black shales, commonly illite). This fixation is believed due to presently undefined chemical processes related to organic fractions in sediments. A similar treatment pertaining to Ge, V, Mo, and other trace elements is possible.
Marine phosphate deposits are probably formed from the dissolved weathered products of igneous rocks transported to sea whereby they are fixed continuously by marine organisms, which after death settle to the sea floor, decay, and aid in the formation of calcium phosphate deposits (Habashi, 1962). Trace amounts of uranium present in water are then absorbed by fine-grained phosphate material and become incorporated into the crystal structure of phosphatic material (e. g. apatite) by proxying for calcium.
Inasmuch as pH and Eh of sea water only permit presence of uranyl ion (primarily as carbonate complex), the U+6 ion (ionic radius = O.80 Å) cannot substitute for Ca (radius = O.99Å). However, U+4 (radius = O.97Å) can substitute for the Ca+2, but the conditions for substitution require an environment capable of reducing [UO2(CO3)3]-4 to U+4. Such conditions prevail where biogenic material predominates, resulting in the complete utilization of dissolved oxygen and thereby assuring a low Eh environment. That such isomorphous substitution occurs is shown by the fact that most U in phosphates exists as U+4. This substitution explains why U content is normally proportional to P2O5 content; as the amount of apatite present increases, the greater the probability of isomorphic substitution.
The uptake of uranium from solution is influenced by many factors, one of which is reduced ionic charge which results from the formation of ion-pairs with anions in solution. Uranium in reduced form (U+4) forms sulphate complexes, (USO4)+2 and U(SO4)2, which have a larger diameter than the uncomplexed U+4 ion, consequently these complexes cannot fit the apatite crystal lattice, leaving Ca+2 to substitute for U+4. Sulphates in solution can enter the crystal lattice by substitution, or under saturation conditions precipitate CaSO4. It has been shown that the more sulphate present in marine sediments, the less the uranium content because of the aforementioned complexing action. Both the Eudora (Lansing Group) and Heebner (Shawnee Group) Shales (and others) of the Upper Pennsylvanian cyclothems of eastern Kansas (Moore and others, 1951) should provide excellent opportunities to test these ideas.
Excepting uranium tied up in the clay or organic fraction of sandstones, the amount actually present with the sand is low in most cyclic deposits. Coatings of carnotite are, of course, notable exceptions.
The preceding discussion briefly describes on a qualitative basis what can be expected concerning the absence or presence (and relative amounts) of uranium to be found under different environmental conditions. Similar descriptions can be presented for anyone of the many trace elements normally encountered or expected in the different lithologic units.
What we have attempted to do is focus attention upon the possible presence of trace-element cycles, correlative with the "normal" periodicity of cyclic sedimentary deposits. Uranium was used to illustrate the relative variations that might be expected.
Although a given trace element, of itself, probably will not be of value, grouping of trace elements such as Cu, Zn, Pb, Mo, V, and Ni can probably furnish valuable information concerning probable physicochemical conditions prevailing in a given environment. Although the approach used here is admittedly a gross over simplification, it does serve both to illustrate an approach to the possibility of delineating trace-element cycles correlative with the different lithologic units and also to emphasize the dearth of information concerning distribution of trace elements within the separate fractions contributing to the make-up of any rock unit.
It is imperative, then, that we obtain considerably more information on the distribution of trace elements in these separate fractions before we can hope to form a clear understanding of any paleoecologic setting.
In closing, I would like to pose the following questions: What are the parameters controlling fractionation and distributional patterns of trace elements in recent sediments? Is fixation by shell-bearing organisms of greater importance than fixation by absorption and ion exchange phenomena acting in the sediments? In what specific forms are trace elements present (i.e. what complexes are most important in fixing the different trace metals)? Is the distribution of given trace elements the same in corresponding members of one megacyclothem? Are they the same from one megacyclothem to another? These are only a few of the questions that require study (and answers) before the application of the concept of trace-element periodicity becomes a practical, detailed, interpretative tool in the study of environmental conditions prevailing during deposition of the cyclic deposits common in the geologic column.
Abelson, P. H., 1959, Geochemistry of organic substances, in Researches in Geochemistry: John Wiley & Sons, New York, p. 79-103.
Bailer, J. C. (ed.), 1956, The chemistry of coordination compounds: Rheinhold, New York, 834 p.
Bell, K. G., 1954, Uranium and thorium in sedimentary rocks, in Nuclear geology: John Wiley & Sons, New York, p. 98-114.
Bowen, V. T., and Sutton, Doris, 1951, Comparative studies of mineral constituents of marine sponges I., The genera Dysidae, Chondrilla, Terpios: Jour. Mar. Research, v. 10, p. 153-167.
Carrol, Dorothy, 1959, Ion exchange in clays and other minerals: Geol. Soc. America Bull., v. 70, p. 748-780.
Degens, E. T., Prashnowsky, A., Emery, K. O., and Pimenta, J., 1961, Organic materials in recent and ancient sediments, part II, Amino acids in marine sediments of Santa Barbara Basin, California: N. Jb. Geol. Palaont., Mh., no. 8, p. 413-426.
Drozdova, T. V., and Yemelyanova, M. P., 1960, Intracomplexes of copper with humic acids: Doklady Acad. Sci. USSR., Earth Sciences Sec., v. 134, p. 245-248.
El Wakeel, S. K., and Riley, J. P., 1960, Chemical and mineralogical studies of deep-sea sediments: Geochim. et Cosmochim. Acta, v. 25, p. 110-146.
Fukai, R., and Meinke, W. W., 1962, Activation analyses of vanadium, arsenic, molybdenum, tungsten, rhenium, and gold in marine organisms: Limnol. and Ocean., v. 7, p. 186-200.
Garrels, R. M., 1960, Mineral equilibria at low temperature and pressure: Harper & Bros., New York, 254 p.
Gehman, H. M., 1962, Organic matter in limestones: Geochim. et Cosmochim. Acta, v. 26, p. 885-897.
Goldberg, E. D., 1957, Biogeochemistry of trace metals: Geol. Soc. America Mem. 67, v. 1, p. 345-358.
Goldberg, E. D., 1961a, Marine geochemistry: Ann. Rev. Phys. Chem., v. 12, p. 29-48.
Goldberg, E. D., 1961b, Chemical and mineralogical aspects of deep-sea sediments, in Physics and Chemistry of Earth: v. 4, p. 281-304.
Gurd, F. R. N., and Wilcox, P. E., 1956, Complex formation between metallic cations, proteins, peptides, and amino acids: Advanc. Prot. Chem., v. 11, p. 311-327.
Habashi, Fathi, 1962, Correlation between the uranium content of marine phosphates and other rock constituents: Econ. Geology, v. 57, p. 1081-1084.
Hirst, D. M., 1962, The geochemistry of modern sediments from the Gulf of Paria, II, the location and distribution of trace elements: Geochim. et Cosmochim. Acta, v. 26, p. 1147-1187.
Hood, D. W., 1963, Chemical oceanography, in Ann. Rev. Ocean. and Mar. Biology: v. 1, p. 129-155.
Hood, D. W., 1960a, Tungsten in bottom deposits of Okhotsk Sea: Doklady Acad. Sci. USSR., Earth Sciences Sec., v. 131, p. 242-244.
Isayeva, A. B., 1960a, Tungsten in bottom deposits of Okhotsk Sea: Doklady Acad. Sci. USSR., Earth Sciences Sec., v. 131, p. 242-244.
Isayeva, A. B.. 1960b, Molybdenum in the deposits of Okhotsk Sea: Doklady Acad. Sci. USSR., Earth Sciences Sec., v. 131, p. 249-252.
Kleinburg, J., Argersinger, W. J., and Griswold, E., 1960, Inorganic Chemistry: D. C. Heath & Co., Boston, p. 211-267.
Krauskopf, K. B., 1955, Sedimentary deposits of rare metals: Econ. Geology, 50th Anniv. Volume, pt. 1, p. 411-463.
Krauskopf, K. B., , 1956, Factors controlling concentrations of thirteen rare metals in sea water: Geochim. et Cosmochim. Acta, V. 9, p. 1-32.
Krumbein, W. C., and Garrels, R. M., 1952, Origin and classification of chemical sediments in terms of pH and oxidation-reduction potentials: Jour. Geology, v. 60, p. 1-33.
Lamb, C. A., Bentley, O. G., and Beattie, J. M. (eds.), 1958, Trace Elements: Academic Press, New York, 410 p.
Levorsen, A. I., 1954, Geology of petroleum: Freeman and Co., San Francisco, p. 1-703.
Martell, A. E., and Calvin, M., 1952, Chemistry of metal chelate compounds: Prentice-Hall, New York, ch. 1-7.
Moore, R. C., 1936, Stratigraphic evidence bearing on problems of continental tectonics: Geol. Soc. America Bull., v. 47, p. 1785-1806.
Moore, R. C., Frye, J. C., Jewett, J. M., Lee, Wallace, and O'Conner, H. G., 1951, The Kansas rock column: Kansas Geol. Survey Bull. 89, p. 1-132. [available online]
Nicholls, G. D., Curl, H., and Bowen, V. T., 1959, Spectrographic analysis of marine plankton: Limnol. and Ocean., v. 4, p. 398-408.
Noddack, I., and Noddack, W., 1939, Die Haufigkeiten der Schwermetalle in Meeres Tieren: Arkiv. Zool., v. 32A, no. 4, p. 1-35.
Osterberg, Charles, 1962, Fallout radionuclides in euphausiids: Science, v. 138, no. 3539, p. 529-530.
Osterberg, Charles, Kulm, L. D., and Byrne, J. V., 1963, Gamma emitters in marine sediments near the Columbia River: Science, v. 139, no. 3558, p. 916-917.
Ostrom, M. E., 1957, Trace elements in Illinois Pennsylvanian limestones: Illinois Geol. Survey Circ. 243, p. 1-34.
Prashnowsky, A., Degens, E. T., Emery, K. O., and Pimenta, J., 1961, Organic materials in recent and ancient sediments, part I, Sugars in marine sediments of Santa Barbara Basin, Calif: N. Jb. Geol. Palaont., Mh., no. 8, p. 400-413.
Rankama, Kalervo, and Sahama, Th. G., 1950, Geochelnistry: Univ. Chicago Press, Chicago, 912 p.
Ronov, A. K., 1956, The chemical composition and the conditions of formation of Paleozoic carbonate layers of Russian Platform, based on the data of lithologic geochemical maps: Doklady Acad. Sci. USSR., Earth Sciences Sec., v. 51, p. 47-50.
Runnels, R. T., and Schleicher, J. A., 1956, Chemical composition of eastern Kansas limestones: Kansas Geol. Survey Bull. 119, pt. 3, p. 81-103. [available online]
Schubert, J., 1954, Interactions of metals with small molecules, in Chemical specificity in biologic interactions: Academic Press, New York, p. 114-163.
Shaw, W. H. R., 1960a, Studies in biogeochemistry--I, a biogeochelnica! periodic table. The data: Geochim. et Cosmochim. Acta, v. 19, p. 196-207.
Shaw, W. H. R., 1960b, Studies in biogeochemistry--II, discussion and references: Geochim. et Cosmochim. Acta, v. 19, p. 207-215.
Swain, F. M., 1961, Stratigraphic distribution of furfurrals and amino compounds in Jurassic rocks of Gulf of Mexico region: Am. Assoc. Petroleum Geologists Bull, v. 45, p. 1713-1720.
Vine, J., and others, 1958, The role of humic acids in the geochemistry of uranium: 2nd U. N. Int. Conf. Peaceful Uses Atomic Energy, p. 15-77.
Vinogradov, A. P., 1953, The elementary chemical composition of marine organisms: Sears Foundation for Marine Res., Mem. 2, Yale Univ., New Haven, 647 p.
Kansas Geological Survey
Comments to webadmin@kgs.ku.edu
Web version July 2003. Original publication date Dec. 1964.
URL=http://www.kgs.ku.edu/Publications/Bulletins/169/Angino/index.html