

Kansas Geological Survey, Bulletin 152, pt. 1, originally published in 1961
Methods of Chemical Analysis for Carbonate and Silicate Rocks
by Walter E. Hill, Jr., Wanda N. Waugh, O. Karmie Galle, and Russell T. Runnels

Originally published in 1961 as Kansas Geological Survey Bulletin 152, pt. 1.
This is, in general, the original text as published. The information has not been updated. An Acrobat PDF version (2.6 MB) is also available.
Abstract
Standard accepted methods of chemical analysis have been adapted to the analysis of carbonate and silicate sedimentary rock and mineral samples. The sequence of analysis is presented to show how several methods interlock to follow one another in a logical order in a minimum of time. The appendix includes a list of necessary reagents and other basic data for the utilization of these methods in any laboratory analyzing geologic materials.
Introduction
Methods of chemical analysis change in response to new demands for greater accuracy and shorter time for analysis. In addition, variation in concentration of each element from sample to sample requires accurate methods that are adaptable to change. The analytical methods described in this report are used in the Geochemistry laboratory of the Kansas Geological Survey and represent evolutionary experience gained in 12 years of chemical analysis of carbonate and silicate rocks for industrial and research purposes. Numerous requests for our procedures prompted preparation of this report and we hope that it will find wide use in industrial and research laboratories concerned with these materials. The geochemical laboratory is called upon to analyze clay, shale, limestone, sand, sandstone, volcanic ash, salt, gypsum, water, and coal of varying purity. Various minerals, as well as igneous and metamorphic rocks, are analyzed also. Only analytical procedures for carbonate and silicate rocks are described in this report and these materials are, in the main, sedimentary rocks. Igneous and metamorphic rocks also may be analyzed using the methods outlined. However, modifications of the procedures must be made because elemental ratios commonly differ from those found in sedimentary rocks. Trace elements, if not determined separately, may contribute to errors in summation.
Acknowledgments
Many analysts have worked in the geochemistry laboratories since this study began, and the authors are indebted to all. The major contributors were A. C. Reed, J. A. Schleicher, Marjorie Utter, Nancy Hambleton, and H. S. Van Nortwick, who, through long association with the laboratory and the methods in use were able to suggest modifications.
Other workers to whom we are indebted are E. E. McGill, Clinton MacDuffee, Lois Lloyd, R. R. Lloyd, Norman Thompson, Loretta Vorse, and T. C. Waugh.
General Procedures
Sampling
Chemical analyses can be no better than the samples of the materials or rocks to be analyzed. Analyses are performed to give a more comprehensive understanding of the chemical components or the amount and kind of specific impurities in the sample. Unless the sample is representative of the entire unit, little will be gained by chemical analysis.
Where samples are wanted from rocks occurring in stratigraphic beds, channel samples are the most representative. Channel samples are taken from top to bottom of the outcrop, chipping small pieces of the rock so that fragments of rock from all parts of the face along a vertical line are retained. Weathered rock should be cleared away first because the weathered rock is oxidized or leached and is not always of the same composition as the fresh rock. Samples should be placed in a clean container and every effort should be made to keep the sample as clean and free of extraneous material as possible to avoid the necessity of washing the sample before crushing.
Classification of Samples
Samples are classified roughly into three major groups for analysis: (1) carbonate rocks, (2) silicate rocks, and (3) miscellaneous materials, requiring special analyses. The last group is not covered in this paper.
Carbonate rocks include limestone (CaCO3), dolomite [CaMg(CO3)2], and accessory carbonates of strontium, iron, and other elements in small amounts. Admixtures of clay, sand, chert, pyrite, and carbonaceous material also are analyzed as part of the sample and reported as elemental oxides. The analytical methods for carbonates require that a large percentage of the sample must be acid soluble and decomposed at elevated temperatures with the quantitative liberation of carbon dioxide.
Silicates include clay, sand, sandstone, and chert, as well as accessory minerals and contaminants; further, the samples may be mixtures of carbonate and silicate minerals in significant amounts. Silicates require a much more rigorous treatment to decompose or break down crystal structures for the determination of the major constituents. Dissociation by evaporation with hydrofluoric acid or the formation of acid-soluble glass by fusion with mixed calcium and sodium carbonates is necessary. Analysis of the elements after solution follows much the same procedure as for carbonates.
The separation of methods into two groups, carbonate and silicate, in effect gives optional methods for the performance of some determinations. Where mixed carbonate and silicate minerals are to be analyzed, it may be desirable to utilize individual determinations from both outlines of analysis.
Sample Preparation
Samples are crushed by mechanical methods or by hand; care is taken to clean all equipment before crushing each sample. The coarsely crushed material is mixed, then quartered or split in order to reduce the size of the sample. The resultant sample of approximately 100 grams is further crushed and ground in the laboratory to pass a 60-mesh sieve. Although porcelain mortars and pestles are adequate for soft rocks, such as shales and clays, mullite or fused alumina mortar and pestle combinations will give less contamination and speedier. grinding when used on hard limestones, dolomites, sands, and sandstones. Frequent sieving also speeds the grinding process.
Grinding and sieving tend to segregate the hard and soft portion> of the sample. They should be remixed by placing the sample on a rubber sheet and rolling and tumbling the powder. Several minutes of remixing will assure homogeneity of the sample. This is necessary because several separate portions of each sample are to be taken and analyzed for different elements. Any deviation from homogeneity will be directly reflected by poor summation of the individual analyses to either above or below 100 percent.
Methods of Analysis
The analytical methods are outlined in the order of consecutive determinations as shown on the schematic flow-diagram of the methods (Fig. 1). For clarity they are discussed as if for a single sample determination. However, in practice it will be found that a set of analyses (four to eight samples) can be performed with very little more effort than the analysis of a single sample.
Figure 1—Flow diagram showing procedure for chemical analysis of carbonate and silicate rocks.
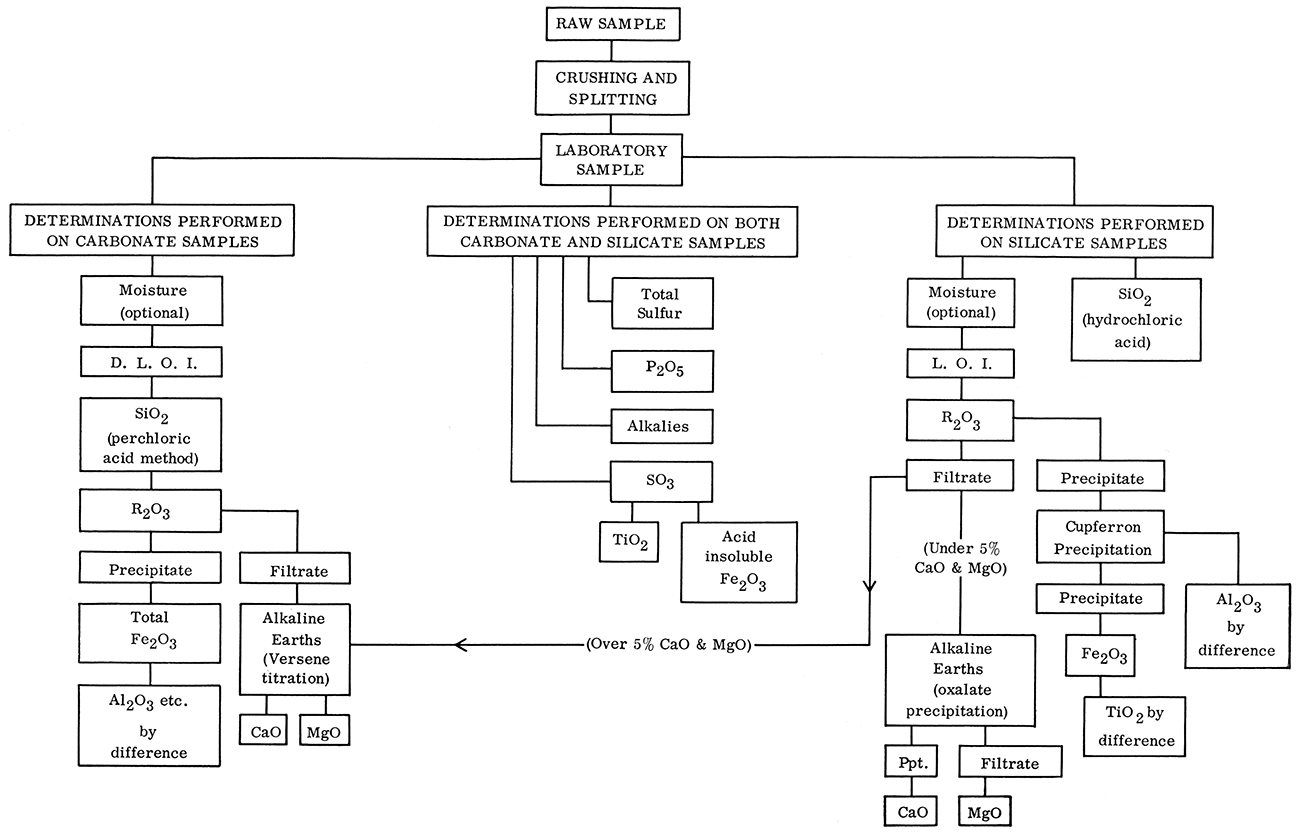
The outline does not show the efficient utilization of equipment or the time gained by performing several determinations consecutively. Delays while evaporating, heating, or igniting will be encountered. These delays offer excellent opportunities to initiate or complete sulfate, sulfide, phosphate, and alkali determinations. Evaporations, some ignitions, and any other steps requiring considerable time should be started in late afternoon if possible in order to utilize the fume hoods, muffles, and hot plates overnight, if operator supervision is not necessary. A set of six samples can be analyzed by one experienced chemist in one and a half to two weeks barring interruptions or problems with the determinations.
Alternate methods, sample calculations, and specific precautions are discussed following the outline of methods.
Procedure for Carbonate Samples
Differential loss on ignition (Galle and Runnels, 1960)
- Weigh approximately 2 g of sample into a clean, tared, platinum crucible. (Record the sample weight for moisture; optional.)
- Dry at 105° C for one hour. Place in a desiccator, cool, weigh, and record the weight at 105° C. (Calculate moisture; optional.)
- Heat an electric muffle, which has an automatic temperature controller, to 550° C. Ignite the sample at 550° C for 25 minutes.
- Remove the sample, cool, and weigh. Calculate the percent loss from 105° C to 550° C:
{[(105° C wt. sample + crucible) - (550° C wt. sample + crucible)] × 100} / 105° C wt. sample = % Loss at 550° C
- Return the sample to the muffle (with a platinum lid on the crucible if the sample is to be left in the muffle overnight). Set the muffle to reach a temperature of 1000° C. Leave the sample in the muffle for at least one hour after the temperature has reached 1000° C.
- Remove, cool in a desiccator, and weigh. Calculate the percent loss from 550° C to 1000° C and record as CO2:
{[(550° C wt. sample + crucible)-(100° C wt. sample + crucible) ) × 100} / 105° C wt. sample = % CO2
Silica (perchloric acid dehydration) (Hillebrand and others, 1953)
- Using distilled water, quantitatively transfer the sample from the D.L.O.I. determination to a 140-ml capacity, black-glazed casserole. Keep the volume of water to a minimum. Add 25-30 ml of 70% perchloric acid. Evaporate on a hot plate until heavy white fumes of perchloric acid are evolved.
- Cover the casserole with a watch glass and increase the hot plate temperature until a steady but gentle refluxing begins. Continue the refluxing for a minimum of 20 minutes. Set the sample off the hot plate and cool at least 10 minutes.
- Add 75 ml of boiling water from a graduated cylinder. Rinse the watch glass with part of this 75 ml of water.
- Stir the sample several times and cool approximately 10 minutes before filtering. Filter through Whatman # 40 filter paper or equivalent into a clean 400-ml beaker. Wash the silica 5 times with boiling 5% hydrochloric acid and then with boiling water at least 10 times. Reserve filtrate and washes for RO2 and R2O3 determination.
- Place the silica and filter paper in the original platinum crucible. Cover with concentrated ammonium hydroxide. Let stand at room temperature overnight. Place the crucible on a hot plate and dry the silica before igniting the sample in the muffle. Ignite very slowly at first in an electric muffle, holding at 400° C until combustion is complete; then proceed to 1200° C and maintain this temperature for 30 minutes.
- Remove from the muffle, cool in a desiccator, and weigh quickly. Record the weight as crude silica.
- To the weighed crude silica carefully add 5 ml of water and 10 drops of 50% sulfuric acid and fill the crucible with 48% hydrofluoric acid. Place on a cold hot plate; slowly increase the temperature to 110° C. Evaporate until only sulfuric acid remains. Slowly increase the temperature until all fumes of sulfuric acid are removed. Place on a Meeker burner, cover with a platinum lid, heat cautiously at first, and slowly increase the heat until at the maximum of the burner; continue heating for one hour. Remove from heat, cool in a desiccator, and weigh. Repeat the heating and weighing steps until a constant weight is obtained. Record this weight as fumed silica residue. Calculate the percent SiO2:
(Crude SiO2)-(Silica residue) = net wt. SiO2
net wt. SiO2 × 100 / dry wt. sample = % SiO2
RO2 & R2O3 group (Fe2O3, Al2O3, MnO2, TiO2) (Hillebrand and others, 1953)
- Fuse the residue from the fumed silica with a small amount (about 2.5 g) of sodium carbonate. Add this fusion quantitatively to the filtrate reserved from the silica determination.
- Add a few drops of bromine water to the sample, place on a hot plate, and boil out the excess bromine.
- Remove the sample from the hot plate. Add a few drops of methyl red and bromthymol blue indicators and a small quantity of macerated filter paper. Add ammonium hydroxide dropwise to a pH of 5.5 to 6.5 (a yellow-green color). The pH can be checked with fresh pHydrion paper.
- Heat the sample to boiling for 2-3 minutes on a burner while stirring to prevent bumping.
- Filter the samples through Whatman # 41 H filter paper or equivalent. Reserve the filtrate in a clean 600-ml beaker.
- Wash the precipitate into the original beaker using a stream or distilled water. Add 5 ml of hydrochloric acid and a few drops of bromine water. Heat to boiling and then repeat the precipitation and filtering process (Steps 3, 4 and 5). Wash the residue at least 10 times with a boiling 2% ammonium chloride solution. Scrub sides and bottom of the beaker with a small piece of filter paper and add it to the main precipitate. Reserve combined filtrates for CaO and MgO determinations.
- Place the washed precipitate in a tared, porcelain crucible. Dry the sample at 200° C and then ignite slowly in an electric muffle to 1200° C and maintain the temperature for 30 minutes.
- Remove from muffle, cool in desiccator, weigh, and calculate percent total RO2 and R2O3:
(wt. RO2 + R2O3) × 100 / dry wt. sample = % (RO2 + R2O3)
Ferric oxide (Hillebrand and others, 1953)
- Gently fuse the RO2 and R2O3 precipitate with approximately 10 g of potassium pyrosulfate over a Meeker burner until solution of the residue is complete.
- Allow the fusion to cool; place the fusion button in a clean 250-ml beaker. Wash the crucible with boiling water and add the washes to the beaker. The total volume of water including the washes should be 100-150 ml.
- Add 10-15 ml of hydrochloric acid; heat the sample carefully over a Bunsen burner until the fusion cake is in solution.
- Add sufficient stannous chloride reagent dropwise until the yellow color of the solution disappears. An excess of stannous chloride is to be avoided. Place the sample in the refrigerator to cool.
- When the sample is below room temperature, titrate with standardized potassium dichromate solution as follows:
- Rapidly add 35 ml of saturated mercuric chloride solution and stir. Let stand 1-3 minutes unless the solution begins to turn milky. If this happens, proceed with Step b immediately.
- Add 15 ml of mixed acid (50-50 sulfuric and phosphoric acid) and 8-10 drops of iron indicator (barium diphenylamine sulfonate), and titrate with standardized dichromate. The end point is a definite change to very deep purple. Record the burette readings before and after titration and calculate as Fe2O3:
(ml of dichromate used × titer of dichromate × 100) / dry wt. of sample = % total Fe2O3
Calcium oxide and magnesium oxide (Versene method) (Patton and Reeder, 1956)
- Add a few drops of bromine water (to remove excess indicator) to the RO2 and R2O3 filtrate, place on a hot plate, and evaporate to approximately 250 ml.
- Filter through Whatman # 40 or Munktells "O" filter paper or equivalent into a 500-ml volumetric flask. Wash with boiling water.
- Cool, make up to volume with distilled water, and shake well.
- Pipette triplicate aliquots (25 ml for most limestones) of the sample into 250-ml Erlenmeyer flasks and dilute to approximately 50 ml.
- To one aliquot add 5 ml of CaO and MgO buffer solution and mix. Then add 30 mg of potassium cyanide and 30 mg of hydroxylamine hydrochloride and mix again.
- Add 0.1 g of total CaO and MgO indicator and titrate from a plum-red color to a pure blue color with standardized Versene solution. The end point is best seen by looking through the solution at a 100-W tungsten light.
- Record the ml of Versene used as the total (CaO and MgO) titration.
- To a second aliquot add 4 ml of 8 N potassium hydroxide, mix, and allow to stand for 3-5 minutes. Then add 30 mg of potassium cyanide, 30 mg of hydroxylamine hydrochloride, and 0.1 g of CaO indicator. Mix and titrate to a pure blue color.
- To the third aliquot add 1 ml less of Versene solution than was required to titrate the CaO alone (Step 8); then add 4 ml of 8 N potassium hydroxide, mix, and let stand for 3-5 minutes.
- Add 30 mg of potassium cyanide, 30 mg of hydroxylamine hydrochloride, and 0.1 g of CaO indicator. Mix and complete the titration. Record the ml of Versene as CaO titration.
- The MgO titration is the difference between the total titration and the CaO titration. Calculate the percent MgO and CaO:
[ml Versene in MgO titration × MgO titer × 20 (aliquot factor) × 100] / dry wt. sample = % MgO
[ml Versene in CaO titration × CaO titer × 20 (aliquot factor) × 100] / dry wt. sample = % CaO
Procedure for Silicate Samples
Loss on ignition (L.O.I.) (Hillebrand and others, 1953)
- Weigh 1 g of sample into a clean, tared, platinum crucible and dry for one hour at 140° C in an oven; reweigh. (Record moisture loss, room temperature to 140° C; optional.)
- Ignite to 1000° C in an electric muffle with an automatic temperature controller for a minimum of one hour. Cool in a desiccator, weigh, and calculate:
{[(140° C weight)-(1000° C wt.)] × 100} / sample wt. at 140° C = % L.O.I.
RO2 and R2O3 group (Fe2O3, Al2O3, MnO2, TiO2)
- To the sample from the L.O.I. determination add 5 ml of water and 25-35 drops of 50% sulfuric acid and fill the crucible with 48% hydrofluoric acid.
- Heat from room temperature to 110° C and evaporate to apparent dryness. Increase the heat slowly to 230° C and hold at that temperature until fuming ceases to remove sulfuric acid.
- When all fumes cease, place the sample over a Meeker burner at low temperature. Slowly increase the temperature until the crucible attains a dull red color. Hold at this temperature for 30-40 minutes.
- Cool; transfer quantitatively into a clean 400-ml beaker. Add 200-250 ml of water, several drops of bromine water, 5 g of ammonium chloride, 1-2 g of macerated filter paper, and 1-2 ml of hydrochloric acid.
- Place the beaker on a hot plate and digest at or near boiling for at least one hour.
- Remove from heat source and add a few drops each of methyl red and bromthymol blue indicators. Add ammonium hydroxide dropwise to a pH of 5.5 to 6.5 (yellow-green color). This end point may be checked with pHydrion indicator paper. Boil the solution with stirring for 1-3 minutes.
- Filter through Whatman # 41 H filter paper or similar quality ashless paper for gelatinous precipitates.
- Wash the precipitate off the paper into the original beaker. Add 5 ml of hydrochloric acid, enough water to make total solution of 200-250 ml, and several drops of bromine water; boil and reprecipitate.
- Filter through the same paper. Wash the precipitate with boiling 2% ammonium chloride. This wash is to be used at least 15 times, in small volumes each time, allowing the precipitate to drain between washes. Reserve the combined filtrates for CaO and MgO determinations.
- Place the drained precipitate in the original platinum crucible. Dry and ignite at 400° C in an electric muffle to remove the filter paper. Increase the temperature slowly to 1200° C and maintain for 30-40 minutes.
- Cool in a desiccator and weigh as RO2 and R2O3:
{[(wt. ignited ppt + crucible) - (crucible wt.) ) × 100} / dry wt. sample = % total RO2 + R2O3
The precipitate is retained and processed for iron and titanium.
Iron, titanium, and aluminum oxides (cupferron precipitation)
- To the precipitate in the platinum crucible add 10 g of potassium pyrosulfate. Fuse over a low flame of a Meeker burner until complete solution of the precipitate is effected.
- The fusion is then cooled and the fusion button placed in a clean 250-ml beaker. The crucible is washed with boiling water and the washes added to the beaker. The total volume of water including the washes should be 100-150 ml. Add 10 ml of 50% sulfuric acid; heat carefully with stirring to complete solution of the button. Filter quantitatively through Munktells "O" filter paper or equivalent into a clean 400-ml beaker. Wash the filter paper at least five times with boiling water.
- Add 30 ml of 50% sulfuric acid and dilute to 300 ml. Cool to 5° C. Add macerated filter paper and 1 % potassium permanganate solution (dropwise) until a faint but persistent pink color is attained. (Excess potassium permanganate should be avoided.) Stir in a 6% cupferron solution in small increments; 10-30 ml normally is needed to complete the precipitation. The precipitation is complete when the supernatent liquid, after stirring and settling, is clear. The solution is filtered through a double paper composed of Whatman # 40 supported by # 41 H or 5 1/2-cm # 360W S & S, or equivalents. (A blank determination should be made.) Filter on a suction apparatus. Place 1-2 ml of cupferron reagent in the suction flask to check the completion of precipitation. If a brown precipitate appears in the flask, precipitation is not complete and the filtrate must be washed back and precipitation completed by the addition of more cupferron to. the solution. A milky-white precipitate in the flask indicates complete precipitation with cupferron. Apply vacuum only after filtration becomes difficult. Wash the precipitate 15-20 times with small portions of cold 10% hydrochloric acid with 1-2 ml of cupferron solution per liter. The filtrate is discarded.
- After washing place the drained precipitate in a tared, porcelain crucible, dry, heat slowly in an electric muffle, and complete the ignition at 1000° C for one hour.
- Cool, weigh, and record as Fe2O3 + RO2 (TiO2, V2O5, ZrO2).
- Ferric oxide is determined from the ignited cupferron precipitate by the method outlined in the carbonate procedure.
- [(Fe2O3 + RO2) - Fe2O3] × 100 / dry wt. sample = % (TiO2 + V2O5 + ZrO2)
Calcium oxide (oxalate precipitation) (Hillebrand and others, 1953)
- The combined R2O3 filtrate is evaporated to 100-150 ml and passed through a Munktells "O" filter paper or equivalent into a 400-ml beaker. Add 5-10 ml of hydrochloric acid and 5 g of ammonium oxalate.
- The solution is heated on a hot plate to approximately 80° C. Calcium oxalate is precipitated by the dropwise addition of ammonium hydroxide to a bright yellow color with methyl red indicator. Allow the solution and precipitate to stand for one hour, no longer, stirring occasionally.
- Filter through Munktells "OK" or Whatman # 40 filter paper or equivalent into a 600-ml beaker. Wash the precipitate off the filter paper into the original beaker and repeat the precipitation using 1 g of ammonium oxalate. Reserve the filtrate and filter paper.
- The second precipitation can stand as long as necessary, although a two-hour wait has been found to be the optimum time. Filter through the filter paper reserved in Step 3.
- Wash at least 5 or 6 times with 0.1% ammonium oxalate solution. Several additional washes are necessary if an extremely heavy precipitate is obtained. The precipitate should also be washed once or twice lightly with water to remove excess oxalate before titrating. The combined filtrates and washes are reserved for MgO determination.
- Place the calcium oxalate precipitate and the filter paper in the original beaker and add 80 ml of water and 10 ml of sulfuric acid.
- The sulfuric acid solution of calcium oxalate is heated to boiling and titrated to a faint but permanent yellow with standardized eerie sulfate (Kolthoff and Sandell, 1943). A blank should be run to determine the amount of eerie sulfate needed to color the solution:
[(ml ceric sulfate - ml blank) × titer × 100] / dry wt. sample = % CaO
Magnesium oxide (diammonium phosphate precipitation)
- The combined filtrate of the calcium oxalate precipitation is evaporated to dryness. Add 35-50 ml of nitric acid, cover with ribbed watch glasses, and again evaporate to dryness. Repeat if necessary to decompose the oxalate compounds. Then add 35 ml of hydrochloric acid and again take the solution to dryness. Repeat if necessary until all nitric acid and nitrate salts are evolved. Cool and add 50 ml of 5% hydrochloric acid to dissolve the residue. Transfer quantitatively to a clean 250-ml beaker. Total volume should not exceed 150 ml.
- Add 2.5 g of diammonium phosphate and several drops of bromthymol blue.
- Ammonium hydroxide is then added dropwise until a precipitate of magnesium ammonium phosphate begins to form. Ammonium hydroxide is very slowly added until the solution is deep blue in color. The precipitate is allowed to settle for a few minutes to an hour. Then add an excess of 5 ml of ammonium hydroxide per 100 ml. The solution is covered with a watch glass and placed in the refrigerator for a minimum of four hours or overnight.
- The precipitate is filtered through Whatman # 40 filter paper or equivalent if heavy, or Munktells "OK" filter paper or equivalent if extremely light. Do not scrub the beaker. Place the original beaker under the filter; dissolve and wash the precipitate quantitatively through the paper with 5% hydrochloric acid.
- For the second precipitation add approximately 0.1 g of diammonium phosphate and bromthymol blue indicator and repeat the precipitation as in Step 3.
- Filter through the same grade of paper and wash with 5% ammonium hydroxide. Place the precipitate and filter paper in a tared, porcelain crucible; ignite to 1100° C, holding at 400° C to allow for the combustion of the filter paper.
- Cool, weigh, and calculate as MgO:
(wt. magnesium pyrophosphate × .3621 × 100) / dry wt. sample = % MgO
Silica (hydrochloric acid dehydration) (Hillebrand and others, 1953) (Silica determination requires a separate sample.)
- Weigh out approximately 2 g of sample into a tared, platinum crucible. Dry at 140° C, cool in a desiccator, and record the dry sample weight.
- To the sample in the platinum crucible add 1 g of calcium carbonate. If the sample is calcareous omit the calcium carbonate. Add at least 6 g of sodium carbonate and stir with a glass rod or platinum wire until an intimate mixture is obtained. Cover the fusion mixture with a thin layer of sodium carbonate. Cover the crucible with a platinum lid and place either on a burner or in an electric muffle. Slowly increase the heat until fusion of the carbonates is obtained. When using a burner, if the cake tends to swell out of the crucible, heat from the top with another burner. If an electric muffle is used, after slowly increasing the temperature hold at 1000° C for one hour. The fusion is heated until all bubbles have been evolved and the resultant mixture is quiet and completely molten. Allow the sample to cool.
- The cake is tapped out of the crucible into a 250-ml casserole. Add water to cover the fusion button. Cover with a large watch glass and carefully add 50 ml of hydrochloric acid under the edge of the watch glass. The crucible should be filled with 5% hydrochloric acid until the remnants of the cake loosen. Quantitatively add this solution and any solids to the casserole. (Reserve the crucible for later ignition.)
- After solution of the fusion button place the casserole on a hot plate. The cover glass should be rinsed and removed and the solution evaporated to dryness. The preparation of the sample should be timed to begin so that overnight evaporation can be utilized. Approximately 15 hours is necessary to completely evaporate the solution and dehydrate the silica. The dry residue in the casserole should not have more than a very faint odor of hydrochloric acid.
- Cool the casserole containing the desiccated silica. Add 25 ml of hydrochloric acid under a cover glass. Reflux at a low temperature on the hot plate for 5-10 minutes. Add 90-100 ml of boiling water; stir until most of the sodium chloride and other soluble salts are in solution. Quantitatively filter through Whatman # 40 filter paper or equivalent and reserve the filtrate. Wash the silica 10 times with boiling 5% hydrochloric acid and then 5 times with boiling water. Place the residue and paper in the original platinum crucible.
- Return the filtrate to the casserole, evaporate, and dehydrate. Filter as before. Wash at least 5 times with boiling 5% hydrochloric acid and a minimum of 10 times with boiling water. The filtrate is discarded.
- Place the second silica residue with the first in the platinum crucible. Ignite very slowly at first in an electric muffle, holding at 400° C until combustion is complete; then proceed to 1200° C and maintain this temperature for 30 minutes.
- Remove, cool in a desiccator, weigh, and record the weight as crude silica.
- To the weighed crude silica carefully add 5 ml of water and 10 drops of 50% sulfuric acid and fill the crucible with 48% hydrofluoric acid. Place on a cold hot plate; slowly increase the temperature to 110° C. Evaporate until only sulfuric acid remains. Slowly increase the temperature until all fumes of sulfuric acid are removed. Place on a Meeker burner, cover with a platinum lid, heat cautiously at first, and slowly increase the heat until the maximum of the burner is attained; continue heating for one hour. Remove from heat, cool in a desiccator, and weigh. Repeat the heating and weighing steps until a constant weight is obtained. Record this weight as fumed silica residue. Calculate the percent SiO2:
Crude SiO2 - Silica residue = net wt. SiO2
(net wt. SiO2 × 100) / dry wt. sample = % SiO2
Procedure for Minor Constituents Determined on Either Carbonate or Silicate Samples
Sodium and potassium oxides (flame photometer determination) (Elving and Chao, 1949)
- Weigh a sample (0.5-3g) in a platinum crucible (so that the sample in Step 7 contains no more than 50 ppm of either alkali) . Dry at 105° C for carbonates, 140° C for silicates, and record the weight. (Ignite all samples containing carbonates at 1000° C for one hour.)
- Add one-third crucible of water and 20-35 drops of 50% sulfuric acid and fill the crucible with 48% hydrofluoric acid.
- Heat cautiously on a small hot plate until a maximum temperature of 110° C is reached. Evaporate to apparent dryness. Increase temperature slowly to 230° C and hold at that temperature until fuming ceases to remove sulfuric acid.
- When all fumes cease, place over a Meeker burner at low temperature. Slowly increase the temperature until the crucible attains a dull red color. Hold at this temperature for 30-40 minutes.
- Cool, transfer quantitatively into a 400-ml beaker, and add 200 ml of water. Digest near boiling for approximately one hour.
- Add macerated paper, a few drops of bromthymol blue, and 5% ammonium hydroxide until a blue color is obtained.
- Filter into a 500-ml volumetric flask through Whatman # 41 H filter paper or equivalent. Wash copiously with hot water. Cool, make up to volume, and shake well. Determine K2O and Na2O on flame photometer following instructions for the specific flame photometer used.
Sulfate (as SO3) (Kolthoff and Sandell, 1946)
- Weigh 3 g of sample (or a sample of appropriate size containing not more than the 0.1 g of sulfur) into a clean 250-ml beaker.
- Add 40 ml of water, cover with a watch glass, and slowly add 10-15 ml of hydrochloric acid. When effervescence ceases, remove and rinse the watch glass. Evaporate the solution to dryness.
- Cool and add 10 ml of hydrochloric acid; cover with watch glass and place on the hot plate to reflux for five minutes.
- Dilute the solution with 90 ml of boiling water. Add aluminum powder to the boiling solution in small increments until the yellow color disappears. (Avoid an excess of aluminum.)
- Filter quantitatively into a clean 600-ml beaker through Munktells "OK" filter paper or equivalent. Wash the residue with boiling water 6-8 times.
- (Reserve the residue from carbonate samples for the acid-insoluble iron and titanium determinations.) Dilute the filtrate to approximately 500 ml.
- Heat to boiling and slowly add 10 ml of 10% barium chloride solution with constant stirring.
- Digest at a moderate temperature for approximately one hour; then let stand overnight.
- Filter through Munktells "OK" filter paper or equivalent and wash 8-10 times with boiling water. Discard the filtrate.
- Place the residue in a tared, porcelain crucible. Ignite carefully to remove the filter paper and complete ignition at 700° C.
- Cool, add 2-3 drops of 50% sulfuric acid, and place on a hot plate to fume until all the fumes of sulfuric acid are expelled.
- Ignite the sample again at 600°-700° C for 30 minutes.
- Cool in a desiccator, weigh, and calculate percent SO3.
{[(wt. crucible + wt. BaSO4) - (wt. crucible)] × .343 × 100 / total wt. sample} = SO3
Acid-insoluble iron and titanium (TiO2 and Fe2O3)
- Place the residue from the sulfate determination in a clean platinum dish and ignite over a Meeker burner until the filter paper is removed.
- When the ignited residue is cool, add approximately 10 ml of water, 7-10 drops of sulfuric acid, and approximately 20 ml of hydrofluoric acid.
- Place on a hot plate and hold at 110° C for at least eight hours. Slowly increase the temperature until all fumes of sulfuric acid are removed.
- Remove the sample from the hot plate, add approximately 10 g of potassium pyrosulfate, and fuse over a Meeker burner until complete solution is effected.
- Quantitatively transfer the cooled fusion button to a 250-ml beaker. The total volume of water should be 100-150 ml. Add 10-15 ml of 50% sulfuric acid and heat over a Bunsen burner with stirring until solution is completed.
- Filter through Munktells "O" filter paper or equivalent into a clean 400-ml beaker. Wash 6-8 times with boiling water.
- Proceed with Steps 3 through 7 of the cupferron precipitation outlined in the silicate analysis.
Total sulfur (bromine-nitric acid method) (Kolthoff and Sandell, 1946)
- Weigh 3 g of sample into a clean, dry, 250-ml Erlenmeyer flask.
- Add 15 ml of (4-1) carbon tetrachloride-bromine mixture. Cover with a watch glass and let stand for 30 minutes.
- Add slowly 20 ml of nitric acid and let stand at room temperature for 30 minutes or until reaction ceases, whichever is longer.
- Evaporate to dryness on a hot plate.
- Add 35-40 ml of hydrochloric acid and again take to dryness. Repeat as necessary to remove all traces of nitrates. Add hydrochloric acid and treat as in sulfate determination beginning with Step 4.
[(% total sulfur as SO3) - (% sulfate as SO3); × .4004 = % S
Phosphate (Kassner and others, 1948)
- Weigh 3 g of sample (or a sample of appropriate size containing not more than 0.4 g of P2O5) into a clean 250-ml beaker. Cover; add 40 ml of water and 15 ml of nitric acid. Digest near boiling for an hour; then bring to a boil.
- Cool, filter through Munktells "OK" filter paper or equivalent, and wash with nitric acid-ammonium nitrate wash solution in small amounts to keep the total volume as near 75 ml as possible. Discard the residue.
- Add 100 ml of ammonium citromolybdate reagent and bring to a boil with caution. Let stand overnight.
- Using vacuum, filter through tared, fritted crucibles. Wash five times with nitric acid-ammonium nitrate wash solution; then wash twice with small amounts of water. Remove from vacuum and wash the bottom of the crucible with water.
- Dry at 105° C for 1-2 hours. Cool, weigh, and calculate as P2O5:
[wt. phosphorus complex × 0.0378 × 100] / wt. sample = % P2O5
Alternate Methods
The differential loss on ignition (D.L.O.I.) determination used in the carbonate analysis (Fig. 1) may be used, instead of the total loss on ignition determination, in the silicate analysis to determine an approximate value for carbon dioxide.
The perchloric acid dehydration determination for silica in the carbonate analysis assumes a low silica value in relatively pure limestone and is very accurate for small quantities of silicon dioxide. If silica values in excess of 20 percent are anticipated, the carbonatefusion and hydrochloric acid-dehydration method described in the silicate analysis should be substituted because of it's higher tolerance for silica variation. This will necessitate using a separate sample for the silica determination.
The R2O3 determination and the iron determination are essentially the same in both procedures. Cupferron precipitation for titanium dioxide may be performed on the residue from the sulfate determination if a titanium dioxide value is desired on carbonate samples. This will also provide an acid-insoluble iron value.
Calcium determination by either oxalate or Versene methods will handle a wide range of percentages with proper dilution and aliquots. The Versene method is simpler and faster but aliquots must be adjusted to handle wide percentage variations of calcium. The Versene method also gives a value of magnesium.
The oxalate method can also be used on a wide range of calcium percentages by careful adjustment of volumes and aliquots, but it is time consuming and does not give a value for magnesium. It is of value primarily in determining low concentrations of calcium in silicate samples directly from the R2O3 filtrate, where making up to volume and aliquoting would create too great a dilution.
Similarly, the separate determination of magnesium is primarily of value for samples containing small quantities of magnesium but the method is sufficiently accurate with appropriate aliquots for large quantities.
Specific Cautions, Sources of Errors, and Calculations
The quantitative chemical analysis of a specific rock or mineral as outlined in this paper may be applied to most samples analyzed. However, some samples require special handling in making a successful analysis. Certain precautions, which provide an accurate analysis with a minimum of work and error, have been developed for these samples. The following section outlines some of these precautions and points out possible sources of error. Also included are a few basic calculations to aid further in providing accurate analyses. The following cautions are included for the less-experienced chemist:
- All water used in analytical procedures is pure distilled water and should be checked at intervals for total solids.
- All evaporations of water and acids should be carried out in a fume hood.
- All solutions should be covered with watch glasses when allowed to stand for any length of time.
- All acids are concentrated unless otherwise specified.
- All glassware and crucibles should be chemically clean.
- All washes are administered from a wash bottle with a fine tip. Wash solutions may be made up in advance and stored in pyrex wash bottles.
Equipment and reagents used in an analytical laboratory govern the accuracy and precision of the results. The equipment should be of high quality but not necessarily the most expensive.
All weighings are made on either one- or two-pan analytical balances capable of weighing in tenths of milligrams. If two balances or more are used, all weights should be calibrated and adjusted to an interchangeable equality, in order to allow the use of either balance through the various steps of the anlysis. Where speed of weighing is a critical factor, chainomatic or direct-reading balances with accompanying air or magnetic dampers are recommended. An open container of a desiccating agent such as calcium chloride or silica gel should be enclosed in the balance case so that the weight of hygroscopic substances will not increase materially while being weighed.
Unless otherwise noted, all glassware referred to is Pyrex-brand glass. Calibrated glassware should meet Bureau of Standards specifications. The laboratory porcelain (casseroles and crucibles) is Coors chemical grade. All chemicals are of analytical reagent grade unless specifically noted.
Although determinations may differ slightly from the accepted analytical procedures used by many laboratories, experience has shown that these methods will produce acceptable results with no sacrifice in accuracy.
The elements are, in most cases, reported as the oxides; this may be inconvenient, but without detailed knowledge of the minerals present few calculations can be made to assign the metallic oxides to the original compounds broken down for analysis. Supplementary D.T.A. and x-ray analyses are invaluable toward this end. Detrimental elements in industrial specifications usually are listed either as percent or in parts per million and for most uses reporting as the oxides is sufficient.
Differential loss on ignition (Galle and Runnels, 1960)
The type of muffle used for this procedure is not important, as there are several commercial types that will produce adequate results. However, it is important that a temperature control unit that will control the temperature within &pplusmn;20° C be used with the muffle. If the temperature is not controlled within this limit, the ignition loss at 550° C will be in error.
The time needed for the loss on ignition at 550° C is a critical part of the determination. The 25 minutes needed to complete the first step of the loss on ignition procedure is a minimum time; with shorter time complete evolution of water and other volatiles and the decomposition of hydrocarbons will not be accomplished. Thirty minutes is the maximum time the sample may be left in the muffle at 550° C. After 30 minutes carbon dioxide begins to evolve and the total carbon dioxide value will be in error.
Assuming the most simple conditions possible, the carbon dioxide value obtained from the loss on ignition may be used to calculate a carbonate value in the following manner:
A sample is analyzed and found to contain in part:
| 41.27% |
CO2 |
| 51.29% |
CaO |
| 1.17% |
MgO |
| 0.05% |
SO3 |
| 0.04% |
P2O5 |
| 93.82% |
|
A small amount of calcium oxide is assumed to be in the form of gypsum and apatite and is calculated as such from the percentages of sulfate and phosphate using these gravimetric factors:
[CaSO4 · 2H2O (MW)] / SO3 (MW) = 2.15013
0.05 × 2.15013 = 0.11 % SO3 as gypsum.
{3 [Ca3 (PO4)2] · CaF2 (MW)} / 3P2O5 (MW) = 2.36846
0.04 × 2.36846 = 0.09% P2O5 as apatite.
CaO (MW) / [CaSO4 · 2H2O (MW)] = 0.32571
0.11 × 0.32571 = 0.04% CaO as gypsum.
10 CaO (MW) / {3 [Ca3(PO4)2] · CaF2 (MW)} = 0.5559
0.09 × 0.5559 = 0.05% CaO as apatite.
The two values for calcium oxide as gypsum and as apatite are then subtracted from the calcium oxide value obtained by analysis to give the value for calcium oxide present as calcium carbonate:
51.29 - 0.04 - 0.05 = 51.20% CaO
Calcium carbonate is then computed from this value:
CaO (MW) / CaCO3 (MW) = 0.5603
51.20 / 0.5603 = 91.38% CaCO3
91.38 - 51.20 = 40.18% CO2 consumed as CaCO3.
41.27 - 40.18 = 1.09% CO2 which is assigned to MgO as MgCO3.
Magnesium carbonate is then computed:
MgO (MW) / MgCO3 (MW) = 0.5219
1.09 / 0.5219 = 2.09% MgCO3
(assumed to be in combination with a small amount of CaCO3 as dolomite.)
In this particular sample there is an excess of 0.17 percent magnesium oxide which is not consumed as magnesium carbonate and may be assigned to the small amount of clay minerals which are indicated to be present by the small percentage of noncarbonate materials determined by analysis.
The calculations outlined above are known to give excellent results on most limestones from the Midcontinent area. However, if the chemical compounds reported by analysis are not consumed in the mineral calculations or the analysis summation is not close to 100 percent, it is possible that apatite and gypsum are not present. The mineral suite should be determined and a proper correction applied.
D.L.O.I. on pyritic limestones (Waugh and Hill, 1960)
The basic procedure for this determination is the same as that for the routine D.L.O.I. However, when limestones contain more than 0.2 percent pyrite a correction is needed for the D.L.O.I. value. The oxidization of pyrite to ferric oxide and sulfur dioxide forms some calcium sulfate at 550° C and causes premature evolution of carbon dioxide. The correction involves determining the amount of sulfate present in the sample, before and after ignition at 1000° C, and the total amount of sulfur present.
Silica (carbonate method)
The black-glazed casserole suggested for this determination contrasts the white color of the silica, simplifying the quantitative transfer of the residue.
If the evaporated perchloric acid solution is not sufficiently cooled before the addition of boiling water, a violent reaction will result. The diluted sample must be cooled to approximately 60° C before filtering or the filter paper will not retain the silica.
After filtration care must be taken to wash the filter paper very thoroughly. Ammonium hydroxide should be added at least once and preferably twice when the washed residue is returned to the crucible. Failure to do this will result in the retention of enough perchloric acid within the filter paper to cause accelerated ignition at approximately 300° C, resulting in the loss of the sample.
Silica (silicate method)
Occasionally tapping may fail to remove the fusion button from the crucible. The sample may be dissolved by placing the crucible in the casserole with the water and hydrochloric acid. After solution remove the crucible and scrub any residue into the casserole.
Make certain that only a trace of hydrochloric acid fumes remains after evaporation so that dehydration of the silica is complete. If the dehydration is not complete some silica will remain soluble.
Where sulfates are present the fumed silica residue from silicate samples may not come to constant weight. Prolonged heating slowly decomposes the sulfates. If a constant weight is not obtained after the heating and weighing process is repeated several times, record the last weight observed as fumed silica residue. Fuse the residue with three to five grams of sodium carbonate. Dissolve the cooled fusion in dilute, hydrochloric acid and proceed with the sulfate determination. The calculated percent silica less the percent sulfate gives the percent pure silicon dioxide.
The sulfate correction can be performed only on silicate samples, because in the carbonate procedure the silica residue must be added to the R2O3 & RO2 group.
R2O3 and RO2 group
Bromine water is added to the solution for R2O3 & RO2 determination to make certain the elements of this group are at their highest oxidation state. The bromine must be expelled before precipitation of the R2O3 & RO2 group begins. Methyl red indicator decomposes in the presence of bromine and, therefore, may be employed as a means of determining the absence of bromine. If the indicator fades, additional boiling of the solution is necessary before ammonium hydroxide can be added.
The pH of 5.5 to 6.5 is critical because aluminum hydroxide is soluble in basic solution and ferric hydroxide is soluble at a lower pH. Ammonium chloride is necessary in the solution to act as a buffer. In the carbonate analysis chloride from the perchloric acid, which is present in the solution, and the added ammonium hydroxide form all the ammonium chloride necessary.
When the R2O3 & RO2 precipitate is large, it may be necessary to use a 12.5-cm instead of an 11-cm filter paper. There should be enough room at the top of the paper to fold the top over the precipitate before placing it in the crucible. Folding the paper over the sample and drying the precipitate prevents sample loss during ignition.
The R2O3 & RO2 precipitate may be ignited in either porcelain or platinum crucibles. If iron is to be determined directly following the R2O3 & RO2 determination, a porcelain crucible must be used. Traces of platinum will titrate with dichromate as iron in a ratio of 10 to 1 platinum over iron. When a cupferron separation is performed between the R2O3 & RO2 and the iron determination, any trace of platinum will be discarded in the filtrate.
In the procedure outlined all of the iron present is reported as ferric oxide and the aluminum oxide is calculated by difference. The aluminum oxide value will contain any manganese dioxide present, and the titanium dioxide value will contain any vanadium oxide and zirconium oxide. Calculation of aluminum oxide is made as follows:
(Total R2O3 + RO2) - [Fe2O3 + (TiO2 + V2O5 + Zr2O3) + P2O5] = Al2O3 + MnO2
Calcium oxide (oxalate)
It is unnecessary to do a double precipitation of calcium oxalate if the calcium oxide is 0.5 percent or less. This can be determined visually because only a very fine, light precipitate will be present.
Phosphate
After filtration of the phosphorus complex the fritted crucible must be washed carefully on the outside or the ammonium nitrate adhering will cause a positive error in the percent phosphate. If the phosphorus complex is dried too long, decomposition will occur (the phosphorus complex changes from bright yellow to green upon decomposition). The fritted crucibles may be cleaned by soaking for a few minutes in ammonium hydroxide.
Sulfate
An excess of aluminum powder should be avoided. Its reaction with hydrochloric acid consumes the acid and the total solution will not be acidic enough for the precipitation of barium sulfate. Aluminum powder in the solution has a tendency to creep and any excess will cause filtration difficulties.
The barium sulfate precipitate may be placed in a crucible that has been used for this determination previously. If the crucible is stored in a desiccator between determinations, the last recorded weight of crucible and barium sulfate may be used as the crucible weight in a succeeding determination. The barium sulfate and wet filter paper should be ignited at a low temperature to prevent reduction of the barium sulfate.
Appendix
Necessary Reagents
Acids:
- Liquid
- Hydrochloric—36.5-38.0%
- Sulfuric—95.0-98.0%
- Nitric—69.0-71.0%
- O-Phosphoric—minimum of 85%
- Perchloric—70%
- Solid
Liquid reagents and solvents:
- 2-Amino-ethanol
- Ethyl alcohol—95%
- Carbon tetrachloride (low sulfur)
- Bromine
- Acetone
Indicators (powdered):
- Barium diphenylamine sulfonate
- Bromcresol purple
- Bromthymol blue
- Cal red
- Eriochrome Black T (F 241)
- Methyl red
- Phenolphthalein
- Thymol blue
Organic solids:
- Benzoic acid
- Cupferron
- Disodium ethylenediamine tetraacetate
- Hydroxylamine hydrochloride
- Phenacetin
Inorganic solids:
- Ammonium chloride
- Ammonium molybdate
- Ammonium oxalate
- Di-ammonium phosphate
- Barium chloride
- Calcium carbonate
- Calcium chloride
- Ceric sulfate
- Mercuric chloride
- Potassium pyrosulfate
- Potassium cyanide
- Potassium dichromate
- Potassium dichromate (Tech.)
- Potassium hydroxide
- Potassium permanganate
- Silica gel (dessicant)
- Sodium carbonate
- Sodium chloride
- Sodium hydroxide
- Sodium oxalate
- Stannous chloride
Metals:
- Aluminum powder
- Iron wire
- Tin
Prepared Reagents
Mixed acid: 50% H2SO4 and 50% H3PO4, by volume. Add cautiously in beaker and let cool.
Mercuric chloride: Saturated solution HgCl2.
Barium chloride: 10 g BaCl2 per 90 ml H2O. Filter and acidify slightly with HCl.
Ammonium citromolybdate (phosphate determination):
- Solution A:
- 93.6 ml NH4OH
- 42.8 ml HNO3
- 1280.0 ml H2O
- 52.6 g citric acid
- 68.0 g ammonium molybdate
- Solution B:
- 253 ml reagent HNO3
- 300 ml H2O
Pour solution A into solution B. Use 2-1 flask. Add handful of glass beads and boil a few minutes with pinch of (NH4)2HPO4. Let stand overnight and siphon into reagent bottle, leaving precipitate in flask. Clean with NH4OH.
Cupferron reagent: 6% solution in water; add very small amount of phenacetin dissolved in few drops C2H5OH to aid in keeping. Store prepared solution in refrigerator. Filter before using or storing.
Stannous chloride: 15 g SnCl2 in 100 ml 1:2 HCl; add metallic tin.
Buffer solution (total CaO and MgO determination): Add 55 ml concentrated HCl to 400 ml distilled water and mix thoroughly. Slowly pour 310 ml redistilled monoethanolamine (2-amino-ethanol) with stirring into the mixture. Cool the solution to room temperature. Titrate 50 ml standard MgCl2 solution with standard EDTA solution using 1 ml monoethanolamine-hydrochloric acid buffer. Use method of total CaO-MgO determination. Add 50 ml MgCl2 solution to the amount of EDTA required to sequester the magnesium exactly; pour the mixture into the monoethanolamine-hydrochloric acid solution and mix well.
Buffer solution (calcium determination only): KOH, 8 N (448.80 g/liter or equivalent), made with reagent grade KOH.
Standard Solutions
N/10K2Cr2O7: Dissolve 4.9 g K2Cr2O7 in distilled water; dilute to 1 l. Standardize against 0.7-g samples of Mohr's Salt or against 0.1-g samples of iron wire.
N/4 Ce(HSO4)4: Dissolve 264 g Ce(HSO4)4 in 500 ml water containing 50 ml H2SO4; dilute to 2 l. Standardize against Na2C2O4.
N/10KOH: 5.61 g of KOH diluted to 1 1. Standardize against 0.4 g benzoic acid.
N/10HCl: 8.55 ml concentrated HCl diluted to 1 l.
Standard CaCl2 solution: Transfer 1 g pure CaCO3 (Spectrographic Standard) to a volumetric flask. Add the equivalent of 250 ml 0.100 N HCl and swirl the contents of the flask until the CaCO3 is dissolved. Add the equivalent of 40 ml 0.100 N KOH to the flask. Let contents cool to room temperature and dilute to a liter.
Standard MgCl2 solution: 2.03 g MgCl2 · 6H2O dried at 90° C and make up a liter of solution. The MgCl2 is fairly hygroscopic and is difficult to weigh with accuracy. A better method would be to dissolve magnesium metal in 0.1 N HCl. The solution should contain 0.4025 mg/ml Mg as MgO. The gravimetric factor from MgCl2 · 6H2O to MgO is 0.19829. The gravimetric factor from Mg to MgO is 1.65789.
N/25 disodium ethylenediamine tetraacetate (Versene): Dissolve 7.8 g in distilled water and dilute to a liter. This solution is standardized against standard CaCl2 solution using Eriochrome Black T as the indicator. (It was found that this concentration of 0.04 N was better suited for our concentrations of CaO than the recommended 0.02 N in the original procedure.)
Wash Solutions
SiO2 (for silicates): Hot 5% HCl
R2O3: Boiling 2% NH4Cl
MgO (pyrophosphate): Cold 5% NH2OH
CaO (oxalate): cold 0.1% NH4C2O4
P2O3: 100 ml HNO3 + 50 ml NH4OH + 850 ml H2O. Mix in flask.
Cupferron: Cold 10% HCl with 1-3 ml 6% cupferron solution per liter.
Indicators
Methyl Red: 1 g powdered reagent in 1 l ethyl alcohol.
Bromthymol Blue: Mix 0.24 g with 150 ml 95% ethyl alcohol. Dilute to 300 ml with H2O.
Phenolphthalein: 1 g in 100 ml 95% ethyl alcohol.
Diphenylamine sulfonic acid: 0.32 g Ba salt reagent, 100 ml H2O, and 3 ml concentrated H2SO4; digest at 100° C. and filter.
Eriochrome Black T (total calcium and magnesium indicator): Triturate 0.2 g Eriochrome Black T and 50 g reagent-grade KCl with a mortar and pestle until all the dye is evenly distributed on the surface of the KCl crystals.
Cal-Red (calcium-indicator): Triturate 0.5 g 2-hydroxy-1-(2-hydroxy-4-sulfo-1-naphthylaxo)-3-naphthoic acid (Cal-Red) and 50 g reagent grade Na2SO4 until the dye is evenly distributed on the Na2SO4.
References
Elving, P. J., and Chao, P. C. (1949) The determination of alkali metals in silicates and similar materials: Anal. Chem., v. 21, no. 4, p. 507-510.
Galle, O. K., and Runnels, R. T. (1960) Determination of CO2 in carbonate rocks by loss on ignition: Jour. Sedimentary Petrology, v. 30, no. 4, p. 613-618.
Hillebrand, W. F., and others (1953, 1955) Applied inorganic analysis: John Wiley and Sons, p. 1-1034.
Kassner, J. L., Crammer, H. P., and Ozier, M. A. (1948) Determination of phosphorus pentoxide in phosphate rock: Anal. Chem., v. 20, no. 11, p. 1052-1055.
Kolthoff, I. M., and Sandell, E. B. (1943, 1946) Textbook of quantitative inorganic analysis: The Macmillan Company, p. 1-794.
Patton, James, and Reeder, Wendell (1956) New indicator for titration of calcium with (ethylenedinitrilo) tetraacetate: Anal. Chem., v. 28, no. 6, p. 1026-1028.
Waugh, W. N., and Hill, W. E., Jr. (1960) Determination of carbon dioxide and other volatiles in pyritic limestones by loss on ignition: Jour. Sedimentary Petrology, v. 30, no. 1, p. 144-147.
Kansas Geological Survey
Placed on web Dec. 11, 2018; originally published July 15, 1961.
Comments to webadmin@kgs.ku.edu
The URL for this page is http://www.kgs.ku.edu/Publications/Bulletins/152_1/index.html