Kansas Geological Survey, Bulletin 152, pt. 5, originally published in 1961

Originally published in 1961 as Kansas Geological Survey Bulletin 152, pt. 5. This is, in general, the original text as published. The information has not been updated. An Acrobat PDF version (6.8 MB) is also available.
Dolomitic carbonates having d-spacing values of disordered dolomite or protodolomite, but lacking major super-reflections, have been precipitated from solutions in the laboratory. Conditions of temperature, pressure, and concentration employed were far less extreme than has previously been reported in the literature. Reagents used were Ca(NO3)2, MgSO4, and Na2CO3; activated charcoal was mixed with reacting solutions to reduce reaction rate so that effect of rate on the final product could be observed.
Experimental results indicate that the pH of the precipitating medium, as controlled by the amount of carbonate ion present, is the most important factor affecting the precipitation of dolomite. Better products were obtained from 1.0M solutions mixed in a Ca++: Mg++: CO3= ration of 1:1:2 where the pH of the precipitating medium ranged from 9.7 at 25°C to 9.1 at 100°C; when the ratio was changed to 1:1:1, other conditions remaining the same, proto-dolomite was the carbonate formed. Increase in temperature of reaction from 25°C to 100°C and in concentration of reagents from 0.1M to 1.0M abet the ordering and crystallinity of the dolomitic precipitate. Reduction in the rate of reaction allowed larger crystals to form; crystal size ranged from about 0.5 to 30 microns.
That the presence of sulfate may be essential to the precipitation of some dolomites was indicated when other salts of magnesium were substituted for the sulfate and the resulting products included no dolomitic carbonate. The genesis of some dolomites, especially those associated with evaporites or evaporative conditions, might require the presence of a natural suspension or gel-like phase.
Vast amounts of petroleum have been and will continue to be produced from dolomitic rocks of Kansas. It is most important that we learn as much as possible about the conditions under which these strata were deposited because of the effect that these conditions might have had on the formation, migration, and accumulation of petroleum in the subsurface reservoirs with which the state has been so generously endowed.
The mineral dolomite has been known as such since 1779 when Arduino named it in honor of the French chemist Dolomieu. Undoubtedly many unreported attempts at the synthesis of this double salt were made previous to 1848 when De Marignac found that a finely ground mixture of calcium carbonate and magnesium chloride, placed in a closed tube and heated, gave dolomite. Further syntheses were generally successful if high temperatures or pressures or both were applied, but met with failure if conditions approaching those of a natural sedimentary environment were simulated.
Of the several generic types of dolomite that have been defined, the most puzzling is the primary chemical precipitate. That this form of the double carbonate could exist was a premise acceptable to most geologists, but it was dogmatically believed that it was a freak of nature, and that very special conditions had to be met before the precipitation took place. These conditions, it was thought, might have existed in the geologic past, but because no dolomite was found precipitating from sea water in the present-day oceans, the uniformitarian principle could not be applied and this geological rarity was to remain an enigma. In 1957, however, Alderman and Skinner reported that dolomite was precipitating from solution in a lagoonal environment in southeastern South Australia. This major discovery lent new impetus to study of the somewhat dormant problem of primary dolomite and of its synthesis in the laboratory. As in the past, however, there was no success in laboratory syntheses unless geologically inconceivable temperatures, total pressures, carbon dioxide partial pressures, or concentrations were employed.
Although the origin and synthesis of primary sedimentary dolomite has been the subject of many studies, the processes that operate in the natural precipitation of that double salt have never been completely understood or integrated. It is, of course, impossible to review every attempt at the synthesis of dolomite in the laboratory; therefore only the major contributions will be noted. Excellent summaries of the early works can be found in papers by Van Tuyl (1916), Cayeux (1935), Fairbridge (1957), and Ingerson (1961).
Among the first reported experiments was that of De Marignac (1848); in his work, a mixture of CaCO3 and MgCl2 was heated in a closed tube at 200°C and 15 atmospheres total pressure, and the resulting product was identified as dolomite. Similar syntheses were affected by Durocher (1851) and Deville (1858) under considerable heat and pressure. Von Morlot (1847) demonstrated that dolomite could be formed by treating a finely crystalline mixture of CaCO3 and MgSO4 with heat and pressure.
The initial attempt at precipitation of dolomite from solution was made in 1859 by Hunt. The precipitate formed by the action of alkaline carbonates with solutions of bicarbonate, however, was not the double salt but rather a mixture of magnesium carbonate and calcium carbonate; these products had to be heated severely before dolomite was obtained. Several variations of Hunt's experiments were tried with different reagents and catalysts (Hoppe-Seyler, 1875; Linck, 1909; Spangenberg, 1913); but the result was the same in these and other experiments because high temperatures or pressures or both had to be applied before dolomite was formed (Clarke, 1924). Obviously the experiments did not hit upon the proper combination or concentration of reactants; more important perhaps is the fact that they did not consider the role played by the partial pressure of CO2 above the solutions.
More recently, syntheses have been made by Chilinger (1956), Graf and Goldsmith (1956), Medlin (1959), Baron and Favre (1959), and Baron (1960). In these works too, however, unnaturally high temperatures, CO2 pressures, or concentrations of reactants were necessary to bring about the crystallization of dolomite from various starting materials.
Most of these experiments have been criticized on the grounds that such artificial conditions of temperature, pressure, and concentration could not exist in nature. Yet no evidence is available to ascertain the extent to which time may replace the artificial conditions of such reactions. It is possible that in the near future such processes may be followed with radioactive tracers so that rates of reaction can be determined even if they are very slow.
In addition to the aforementioned studies, it should be noted that investigations have been and are being made concerning the role of bacteria in dolomite precipitation. Lalou (1957) reported the formation of dolomite under laboratory conditions in a marine aquarium with cultures. Neher and Rohrer (1958) described the precipitation of dolomite from solution by the action of living bacteria that can be isolated and studied under controlled conditions; they hope to determine whether bacterial action is brought about by an enzyme that contains a characteristic trace metal. If some dolomites are found to contain the trace metal consistently and in appropriate amounts, the bacterial origin of such dolomites would be indicated.
The chemical composition of natural waters is complex and rarely known in detail. Solution studies have shown that there is a tendency for simple ions and molecules to interact, even in dilute inorganic solutions, thus forming complex species (Hood and others, 1959; Garrels and others, 1960). These complexes can and probably do make important contributions to the total matter carried in solution. In addition, natural waters contain organic species, some of which are effective for binding metal ions. Salinity, temperature, carbon dioxide partial pressure, total pressure, pH and Eh, all vary greatly in sea water. Validity of calculations concerning carbonate equilibria for natural systems are limited because of these and other variables.
Precipitation of a compound from solution takes place if its solubility product constant (Ksp) is exceeded. The solubility product constant for a given temperature is a measure of ion concentration and applies to saturated solutions of a salt.
The reliability of the interpretations made by use of this principle is determined mainly by the total concentration of ions in solution, and in very dilute solutions (0.001M), calculations are correct within about 4 percent. For more concentrated solutions in which the activity coefficients of the ions are considerably less than unity, the actual solubilities of the salts are usually somewhat larger than calculated (Pauling, 1956).
The Ksp values for geologic material commonly associated with carbonate deposition are presented in Table 1. From these values, we can see that at a given temperature, dolomite is much less soluble than is calcium carbonate; the inference, therefore, is that we should get dolomite as a direct carbonate precipitate in preference to either aragonite or calcite. Because of the aforementioned variable factors, however, especially rate of formation of the carbonates and concentration of magnesium, this does not happen.
Table 1—Solubility products of certain mineral species involved in carbonate deposition. Data from Kramer (1959); Lange and Forker (1956); and Sverdrup, Johnson, and Fleming (1942)
| CaMg(CO3)2 | 2.95 × 10-13 | In sea water at 25°C |
| CaCO3 | 6.5 × 10-5 | In sea water at 25°C |
| Mg(OH)2 | 5 × 10-11 | In sea water at 20°C |
| SrCO3 | 500 × 10-9 | In sea water at 20°C |
| CaSO4 | 6.1 × 10-5 | In distilled water at 18°C |
Thermodynamic studies are used to determine whether a reaction will proceed either by itself or in competition with other reactions in the same system. Such studies do not, however, give any indication of the rate of reaction; rate of reaction is treated by kinetics. The fundamental parameters upon which most thermodynamic and kinetic calculations depend are temperature, pressure, and concentration of reactants. These factors are interrelated and are used to determine the free energy change in a reaction, the free energy of formation of an element or compound, and rates at which elements or compounds form.
Halla (1935, 1936) found that at normal temperature and pressure the dolomite-forming reaction has a negative free energy change and should proceed spontaneously. His thermodynamic calculations indicated that the direction of the reaction was controlled solely by calcium and magnesium ion concentration available as governed by the Law of Mass Action: 2CaCO3 + Mg++ = CaMg(CO3)2 + Ca++; -ΔFR°. He made no observations concerning the rate at which the reaction could proceed.
Values for free energy of formation of materials commonly associated with carbonate deposition are given in Table 2. These values and those for solubility found in Table 1 indicate that of the carbonates—dolomite, aragonite, calcite, and magnesite—dolomite is stable in normal sea water with respect to calcium carbonate or mixtures of calcium carbonate and magnesium carbonate. This fact makes the problem of nonprecipitation of dolomite extremely challenging. A partial answer to this problem might be found in other physical and chemical quantities that have not yet been completely evaluated. Robie (personal communication with Garrels and others, 1960) has determined the heat capacities of dolomite, calcite, and magnesite and is now working on the entropies, heat contents, and heats of formation of these minerals. Barton (USGS, personal communication with Ingerson; 1961) is presently determining the activity coefficient of magnesium ion in solution; although the activity coefficient of calcium is well established, that of magnesium is in great doubt and may differ significantly from its concentration in the sea, thus affecting the solubility product, and establishing a possible "activation energy barrier" to dolomite precipitation.
Table 2—Values of free energies of formation (ΔFR°) at 25°C and 1 atmosphere pressure for species commonly associated with carbonate deposition; c, crystalline; aq, aqueous; g, gaseous. (Compiled from Garrels, 1960).
| Species | State | ΔFR°, kilocalories |
|---|---|---|
| Ca++CO3 | aq | -132.18 |
| CaCO3, aragonite | c | -269.78 |
| CaCO3, calcite | c | -269.53 |
| CaSO4 | c | -315.56 |
| CaSO4, soluble | c | -313.52 |
| CaSO4, soluble | c | -312.46 |
| CaSO4 · 1/2 H2O-α | c | -343.02 |
| CaSO4 · 1/2 H2O-β | c | -342.78 |
| CaSO4 · 2H2O | c | -429.19 |
| CaMg(CO3)2 | c | -519.99 |
| CO2 | g | - 94.2595 |
| CO2 | aq | - 92.31 |
| H2CO3 | aq | -149.00 |
| HCO3- | aq | -140.31 |
| CO3= | aq | -126.22 |
| Mg++ | aq | -108.99 |
| Mg(OH)2 | c | -199.27 |
| MgCO3 | c | -246.00 |
| Na+ | aq | - 62.589 |
| H2O | aq | - 56.69 |
| OH- | aq | - 37.595 |
| Cl- | aq | - 31.350 |
| SO4= | aq | -177.34 |
| NO3- | aq | -26.43 |
Calculations of the free energy of formation for dolomite are found in the Appendix. Free energy data for the reactions proposed in this study for dolomite precipitation are also found in the Appendix. The calculations indicate that the reactions proposed should proceed inasmuch as the free energy values for the coupled reactions range from -161.51 to -192.07. The range in free energy values is offered because the type of calcium sulfate found in the diffraction patterns of the precipitates could not be identified with certainty. Accordingly, two sets of calculations were made, one using the high ΔFR° for soluble anhydrous CaSO4 and one the lower ΔFR° for α-CaSO4 · 1/2 H2O in order to span the range of free energies of the more likely calcium sulfate precipitates.
No quantitative data have appeared in the literature concerning the rate at which dolomite formation or precipitation takes place. Riviere (1939a) has demonstrated that calcium carbonate will react with CO2-free sea water to add 8 mol percent MgCO3 in a year. Kramer (1959) mentions that the probable reason calcium carbonate forms rather than dolomite in sea water is the rate at which the carbonates form. No experimental work other than that of Riviere has been presented.
Although thermodynamics dictates that dolomite precipitation should take place and that dolomite will be a stable product under normal marine conditions, there must be an adequate supply of the mineral-forming elements available at the site of sedimentation.
Ground water generally is low in magnesium, hence the only logical source of magnesium is ocean water, either as it is or in a concentrated form (brine). Detailed chemical analyses show that there is about three times as much magnesium as calcium in sea water, i.e., 1.27 ppt (parts per thousand) Mg vs. 0.40 ppt Ca; expressed in gross figures, 17 x 1014 metric tons Mg vs. 5.5 X 1014 metric tons Ca (Sverdrup and others, 1942). It is also known that calcium is added to the ocean at four times the rate at which magnesium is added. The answer to calcium deficiency lies in the low solubility of calcium salts, particularly the carbonate; it is frequently supersaturated in the ocean and its removal is effected by chemical precipitation. Magnesium, however, never approaches saturation in sea water at normal temperatures, pressures, and pH, and is not removed, thus building up in concentration as calcium concentration remains stable or is decreased by precipitation. It has been proposed, but not yet investigated, that an excess of calcium ions inhibits magnesium deposition. This, together with the lack of magnesium saturation, might. be a clue to the nonprecipitation of dolomite.
Carbonate ion concentration in sea water is about 0.14 ppt. Local conditions such as inflow of river waters, agitation of surface water, photosynthesis and other oxygen molecule reducing reactions, bacterial action, and volcanic activity may increase the concentration greatly.
Mg(OH)2 can be precipitated from sea water by addition of free alkali. In addition, both calcium and magnesium are in delicate equilibrium where slight changes in alkalinity and CO2 partial pressure may cause precipitation. If an increase in alkalinity results from free base alone, the magnesium will be rapidly precipitated as the pH reaches about 10; in lagoonal environments such as that reported by Alderman and Skinner (1957), the natural waters approach a pH = 10, permitting the precipitation of both calcium and magnesium simultaneously. Although CaCO3 is relatively insoluble and MgCO3 is relatively soluble in saline waters, Ca(OH)2 is much more soluble than Mg(OH)2; thus it might be assumed that there would be little chance for true precipitation of dolomite except by the interaction of CaCO3 and Mg(OH)2 in a gel or suspension (Cloud and Barnes, 1946) and that there is less chance of precipitation of pure MgCO3, which, indeed, is not found in marine sediments. A gel, perhaps containing calcium, magnesium, and carbonate ions, precipitated from sea water by the intermediary of organic activity might be subjected to diagenesis after precipitation, and the dolomite-forming process could then be completed.
An examination of dolomite distribution in the geologic rock column allows the premise that magnesium concentration in sea water has not always been the same as it is now. If, for example, the magnesium concentration was much greater, a point of near saturation could have been reached either in sea water or in solutions undergoing evaporation, as is proposed for the Permian Reef area of Texas and New Mexico (Newell and others, 1953). A situation exists now in southeastern South Australia where dolomite is being precipitated from waters in which salinity ranges to four times that of the ocean (Alderman and Skinner, 1957; Alderman, 1959). If laboratory experiments can be made within reasonable salinity limits, it might be possible to determine whether a mode of precipitation effective in laboratory studies could have been operative in past geologic environments.
Increasing CO2 content of natural waters results in lowering of pH, and consequent greater solubility of carbonate phases present; this in turn probably aids dolomite formation in the manner proposed by Halla (1935, 1936) and Riviere (1939a). Conversely, decreasing CO2 content in aqueous environments causes a rise in pH and an increase of carbonate ion concentration, allowing the magnesium ion to coprecipitate with the calcium ion thus forming primary dolomite.
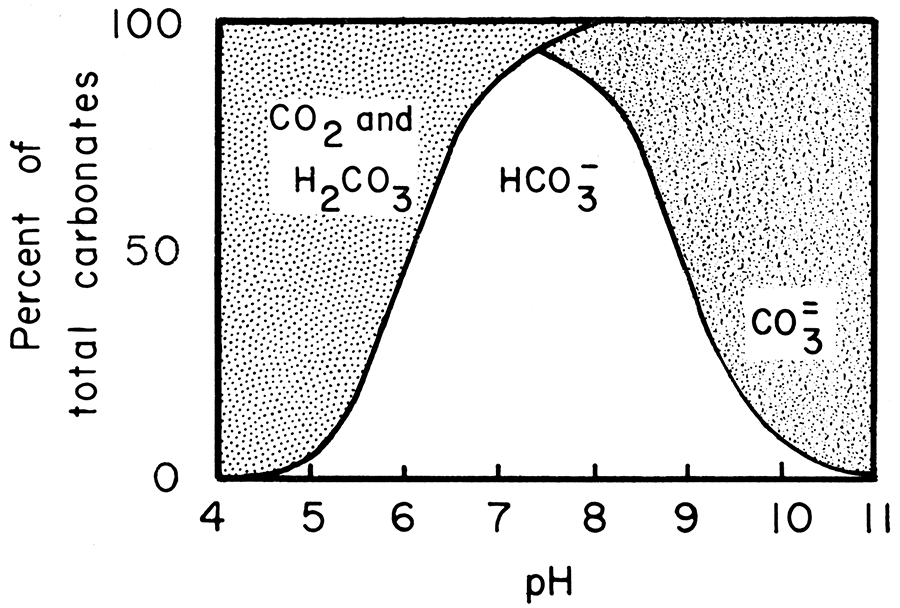
The relationship between pH in sea water and the equilibrium CO2 (dissolved) ↔ H2CO3 ↔ HCO3- ↔ CO3= is shown in Figure 1.Figure 1—Distribution of carbonate species as a function of pH in sea water. (Modified after Weyl, 1961.)
The effect of changing CO2 pressure upon the formation or precipitation of dolomite has not been evaluated. It is known that great changes of CO2 partial pressure cause relatively small changes of pH and would not affect the carbonate ion concentration very much; this is especially true of sea water, which has a self-contained buffering system. The CO2 content of natural waters may be increased or decreased as follows:
| Increase CO2 content | Decrease CO2 content |
|---|---|
| Currents from polar regions Increase of atmosphere pCO2 Respiration Carbonate precipitation Evaporation Increase in depth |
Photosynthesis Bacteria which reduce oxygen-containing molecules Agitation of surface waters Carbonate solution Decrease in salinity |
Chilingar (1956) reported that dolomite was precipitated from evaporating sea water saturated with MgCO3 and CaCO3 at a CO2 pressure of 4 atmospheres; however, Medlin (1959) reported that he could not reproduce Chilingar's experiment. It is unlikely that a supersaturated ocean plus an atmosphere as strongly charged with carbon dioxide as Chilingar indirectly proposed have existed in areas of primary dolomite formation.
Photosynthesis has been called upon many times to account for a lowering of CO2 content of marine environments and a consequent rise in pH. Recent work (Hood and others, 1959) has demonstrated that marine algae use the HCO3- ion rather than the CO2 molecule for photosynthesis; this forces us to reconsider changes that take place in the equilibrium CO2 (dissolved) ↔ H2CO3 ↔ HCO3- ↔ CO3= as regards calculations made on the assumption that CO2 was always the form used in photosynthesis. The effect of this particular factor on solubility and carbonate precipitation has not yet been reported.
Kramer (1959), on the basis of a study of solubility products and free energy data, confirmed suggestions that increased CO2 content of natural waters and consequent smaller carbonate ion concentration would favor the development of dolomite saturation over that of calcite. He further theorizes that formation of calcite predominates over dolomite because of the relatively slow rate at which dolomite forms, although he presents no quantitative data on the rate of carbonate development.
Garrels and others (1960) oppose Kramer's idea and state that the stability of dolomite relative to calcite is not a function of the CO2 content; they infer that a lower partial pressure of CO2 (higher pH and increased carbonate ion concentration) somehow drastically increase the solubility of dolomite relative to calcite.
It is generally accepted that a higher temperature favors the precipitation of dolomite. A higher temperature could affect an aqueous environment in two ways, either of which would aid the precipitation of dolomite. First, increased evaporation would result in an increase in salinity; second, at a higher temperature an aqueous medium would tend to hold less CO2 and there would be a rise in pH and carbonate ion concentration.
Dolomite has been precipitated from solutions in the laboratory under conditions that more nearly approach those of a natural sedimentary environment than do those of experiments previously reported. Physico-chemical parameters that were controlled were temperature, pressure, concentration, and rate of reaction.
The chemicals used in this investigation were of analytical grade and met ACS specifications. A stock solution of calcium ion, one molar concentration (1.0M) , was prepared by dissolving 236.16 grams of calcium nitrate in distilled water and diluting to one liter; a stock solution of magnesium ion, 1.0M, was prepared by dissolving 246.50 grams of magnesium sulfate in distilled water and diluting to one liter; a stock solution of carbonate ion, 1.0M, was prepared by dissolving 105.99 grams of sodium carbonate in distilled water and diluting to one liter. Suitable aliquots were taken from these stock solutions and diluted for use at lesser concentrations.
Solutions to be used at higher than room temperature were heated on a laboratory hotplate, and their temperatures were recorded with standard mercury thermometers to ±2°C.
A Photovolt Electronic pH Meter, Model 125, was used to measure the pH of precipitating media. Measurements are accurate to ±0.1 pH unit.
Rates of reaction in the precipitation could not be quantitatively determined, but activated charcoal was added to the calcium and magnesium solutions to retard the process. Because of its outstanding absorbancy, activated charcoal is an excellent rate reducer and will absorb cations until they react with suitable anions.
100-milliliter (ml) aliquots of calcium nitrate and magnesium sulfate and a 200-ml aliquot of sodium carbonate were taken from the stock solutions. The calcium- and magnesium-bearing solutions were poured into a 600-ml beaker simultaneously and were subjected to mechanical stirring action. Immediately upon the formation of a precipitate, the carbonate solution was added to the mixture, which was then stirred continuously for 15 minutes. The precipitate was next allowed to digest for about 2 hours. After separation from the filtrate, the precipitate was dried and identified by x-ray technique.
To study the effect of rate of reaction on the final products, 1 gram of finely ground activated charcoal was added to each of the calcium- and magnesium-bearing solutions before they were mixed together. To study the effect of temperature, solutions were heated to specified temperatures before precipitation. The stock solutions were diluted to desired concentrations if this parameter was to be studied, but the relative volumes of reagents used remained the same.
X-ray diffraction data were obtained by use of a General Electric XRD-3 proportional counter diffractometer. Chart patterns were run at a scanning rate of 0.2° 2θ per minute so that sufficient detail was available for interpretation. A 1° beam slit, 0.2° detector slit, and nickel-filtered copper radiation were used. The x-ray unit was operated at 48 kv and 15 ma.
The -100-mesh fraction was packed into a plastic holder for use in making the chart patterns.
The National Bureau of Standards Dolomite Sample No. 88 and dolomite patterns presented by Graf and Goldsmith (1956) were used as standards of comparison. Additional identification was made by using calculated d-spacing values and comparing these with d-spacing values presented in the American Society for Testing Materials (ASTM) index file.
Until 1957, when Alderman and Skinner described dolomite precipitating from sea water in a natural environment, many geologists (Twenhofel and others, 1932, p. 342; Merwin, in Twenhofel and others, 1932, p. 339) believed that direct precipitation of dolomite was possible, but hardly probable. It was thought that if the precipitate could form it would have to form in an extremely saline (hypersaline) environment and the product would be associated with evaporites. Early solubility and thermodynamics data, geological field observations, and complete lack of success at producing dolomite from solutions in the laboratory at geologically possible conditions, all served to negate the existence of primary dolomite. Results of experiments made in this investigation indicate that dolomite can form as the direct chemical precipitate under geologically "normal" environments. The effect of physico-chemical parameters controlled in the precipitation experiments and the proposal of reactions and a mechanism for dolomite precipitation are presented in a later section of this report.
Reported attempts at the synthesis of dolomite from solutions in the laboratory have met with failure unless high temperatures or pressure or both were employed. Although it has been observed that primary dolomites are commonly associated with gypsum or anhydrite, this fact was neglected by experimenters, and sulfate salts were not included in experimental precipitating media. The reagents used in the present study include magnesium sulfate together with calcium nitrate and sodium carbonate. The controlled physico-chemical parameters were temperature, pressure, concentration, relative volumes of reagents, and rate of reaction; the effect of these on the products of the precipitation were noted. It was originally planned to make precipitations under increased partial pressures of CO2, but calculations made using pCO2 values considered reasonable for paleogeologic atmospheres indicated that the carbonate ion concentration and pH of precipitating media would not be significantly altered, hence these experiments could be eliminated. The calculations are presented in the Appendix.
The dominant mineral phase present in the precipitates was disordered dolomite, or proto-dolomite of Graf and Goldsmith (1956). Always associated with this phase was some form of calcium sulfate; γ-CaSO4 was the form most commonly identified in the diffraction traces, but α-CaSO4 · 1/2 H2O may also have been present. Additional products observed from the x-ray patterns were calcium carbonate and hydromagnesite; the calcium carbonate was present as either calcite or aragonite, the crystal form being determined by the conditions under which the experiments were made.
Graf and Goldsmith (1956) have defined protodolomite as "single phase rhombohedral carbonates which deviate from the composition of the dolomite that is stable in a given environment, or are imperfectly ordered, or both, but which would transform to dolomite if equilibrium were established." The imperfect ordering is observed in the absence of a superlattice and of the so-called ordering or superlattice reflections at 4.02, 3.72, and 2.53Å (22.1, 23.9, and 35.4° 2θ respectively). These proto-dolomites produce diffuse x-ray reflections and tend to have slightly expanded lattices; however, the general sharpness and position of the reflections can be correlated with the relatively ideal reflections of true dolomite having a well-ordered structure.
In this investigation, the x-ray diffractograms do not show the superlattice reflections reported by Graf and Goldsmith for true dolomite and inasmuch as the major dolomite reflections are diffuse, seemingly owing to disorder, it is believed that the product of crystallization fits the definition of proto-dolomite proposed by Graf and Goldsmith.
A two-step reaction is proposed for the precipitation of dolomitic carbonate as it was performed in this work. The first step results from the addition of 100 ml of magnesium sulfate solution to 100 ml of calcium nitrate solution. The second results from the addition of 200 ml of sodium carbonate solution to the gel or suspension product of the first step. It should be noted that the form of the calcium sulfate that results from the first reaction step could not be identified. Accordingly the general formula CaSO4 is used in the proposed reactions. These reactions, the stoichiometric relations of which are unknown, are presented in Table 3.
Table 3&mdasp;Proposed nonstoichiometric reactions for precipitation of dolomitic carbonate.
| (1) | Ca++ + 2 NO3- + Mg++ + SO4= | → | CaSO4 + Mg++ + 2 NO3- calcium sulfate gel and hydrated magnesium ion gel |
| (2) | CaSO4 + Mg++ + 2 NO3- + 4 Na- + 2 CO3- | → | CaMg(CO3)2 + CaSO4 + 4 Na+ + 2 NO3-
Dolomite |
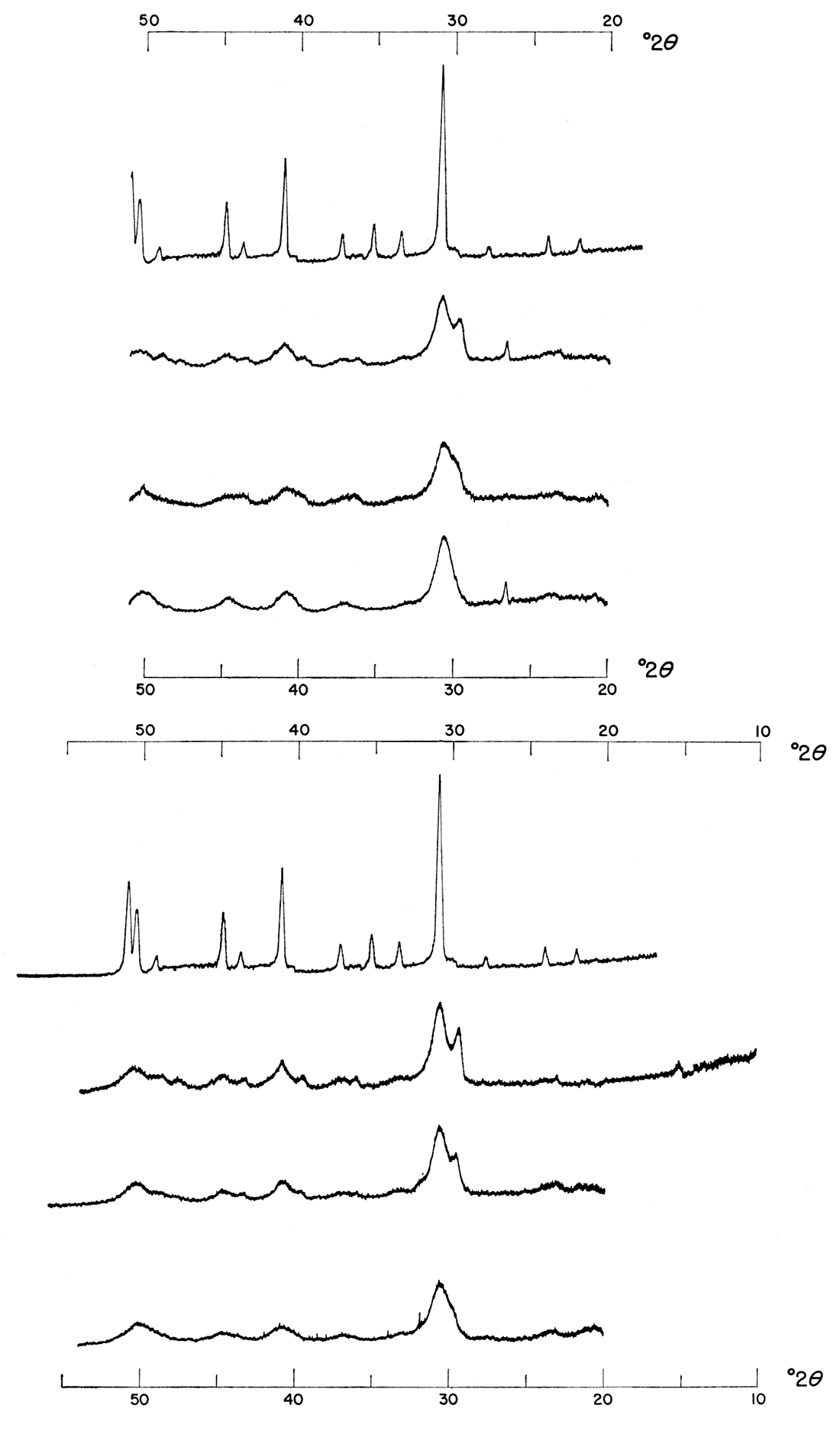
In the precipitation procedure outlined, the relative volume of carbonate ion solution markedly affected the final product. As can be seen in Figure 2, those precipitations made at a 1:1:1 molar concentration ratio of Ca++: Mg++: CO3= gave at best a poorly ordered, expanded lattice type of protodolomite; at 25°C associated products were calcium sulfate and calcium carbonate; at 50°C and 100°C, calcium sulfate was the co-product. The precipitates formed from a 1:1:2 molar concentration ratio, however, gave products exhibiting much better ordering and crystallinity as shown by the position and intensity of comparable major reflections. The precipitates shown in Figure 2 were made in the presence of activated charcoal. The same effect is observed in Figure 3, however, which shows precipitates made without any activated charcoal.
Figure 2—Effect of increasing relative volume of carbonate solution. Precipitates were formed from 1.0 M solutions mixed at various temperatures in the presence of activated charcoal. Precipitates in top of figure were made with a 1:1:1 molar concentration ratio of Ca++: Mg++: CO3=; those in bottom of figure were made with a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3=. Note shift and increase in intensity of major dolomite reflection.
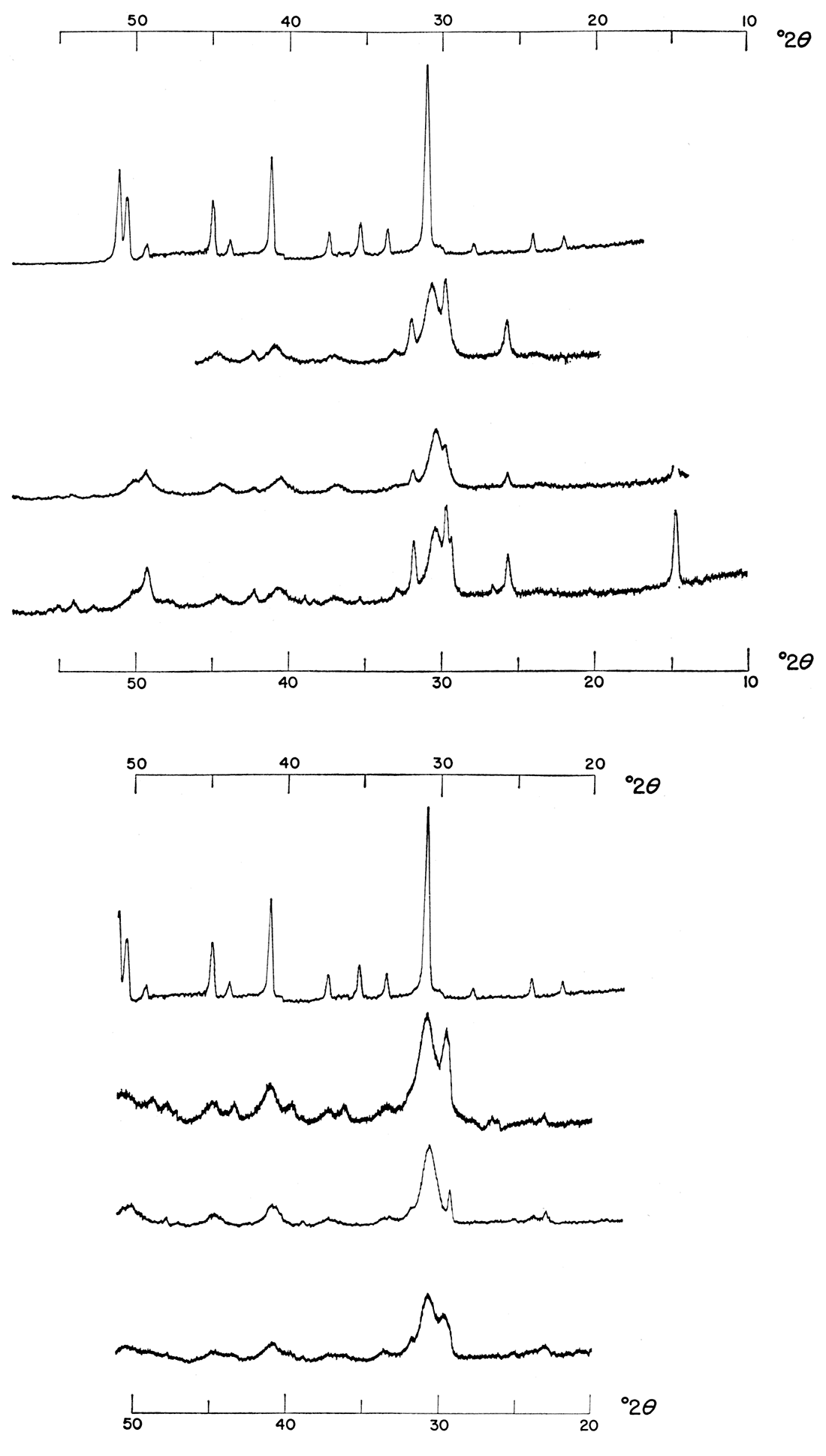
Figure 3—Effect of increasing relative volume of carbonate solution. Precipitates were formed from 1.0 M solutions mixed at various temperatures without activated charcoal. Precipitates in left of figure were made with a 1:1:1 molar concentration ratio of Ca++: Mg++: CO3=; those in right of figures were made with a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3=. Note shift and increase in intensity of major dolomite reflection.
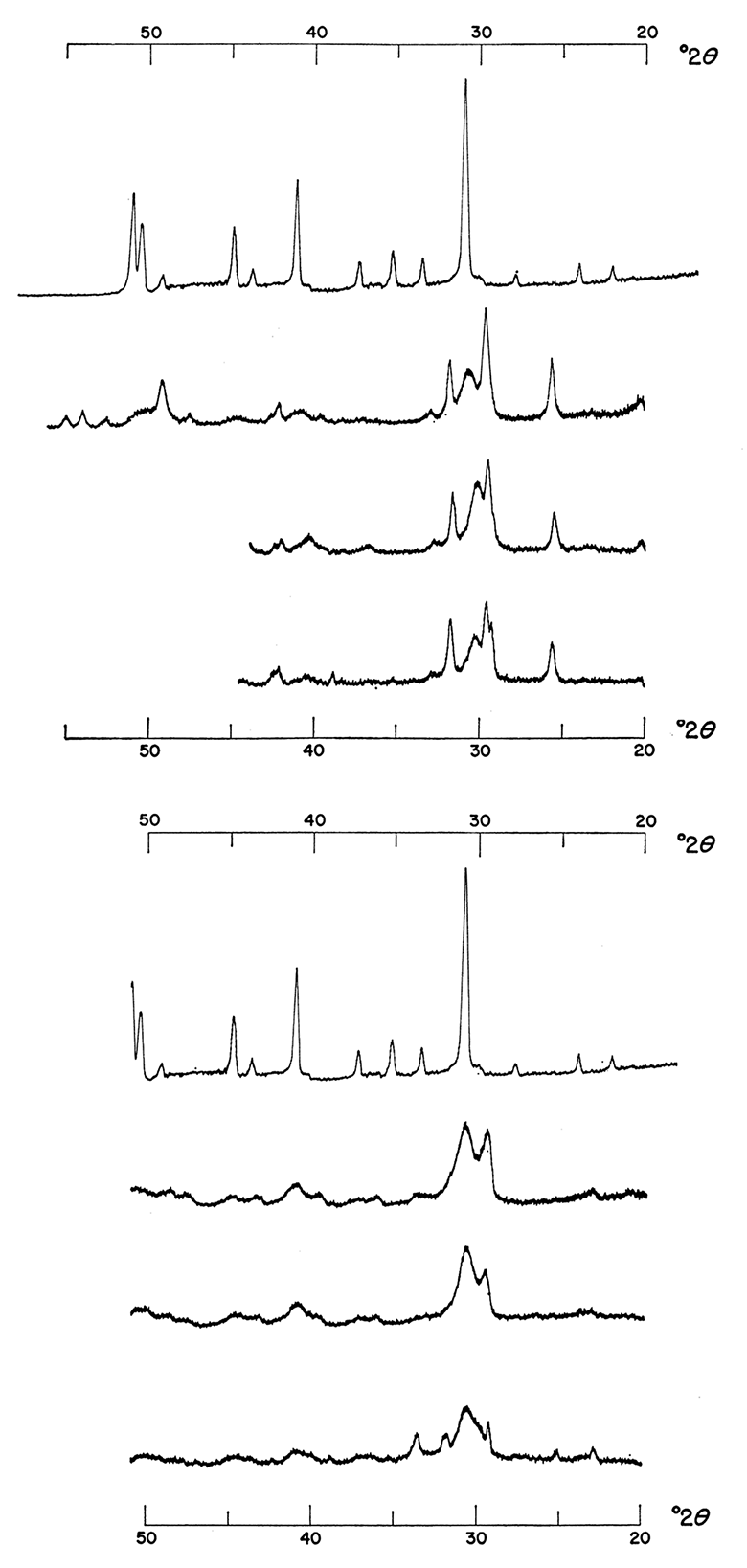
The obvious explanation for this shift from a disordered to a better ordered dolomite lies in the changes in pH as brought about by the increase of carbonate ion of the precipitating medium. At 25°C and a 1:1:1 molar concentration ratio, the pH of the aqueous medium was 8.7 ± 0.1 but at a 1:1:2 ratio of Ca++: Mg++: CO3=, the pH at which precipitates were formed was 9.7 ± 0.1. Similarly, at 100°C and a 1:1:1 ratio, the pH of the aqueous medium was 7.8 ± 0.1 whereas at a 1:1:2 ratio, the pH was 9.1 ± 0.1. The pH values reported from the Australian site of dolomite precipitation were as high as 9.4 (Alderman and Skinner, 1957). It should also be noted that although calcium carbonate is present in the precipitates formed at 25°C it is absent from the precipitates formed at higher temperatures; similarly, at lower pH values calcium sulfate is present in greater quantity. As will be demonstrated, the increase of intensity of the dolomite diffraction maxima is related to rate of reaction and increasing temperature.
For each chart pattern in Figures 2-9, inclusive, the conditions under which the sample was synthesized are listed in Table 4.
Table 4—Experimental and x-ray data. (All experiments were made at atmospheric pressure.)
| Figure | Relative volumes Ca++ :Mg++ :CO3= |
Concentration | Temperature | d-spacing value, major dolomite reflection |
Activated carbon |
|---|---|---|---|---|---|
| 2 | 1:1:1 | 1.0M | 100°C | 2.919Å | Present |
| 1:1:1 | 1.0M | 50°C | 2.938 | Present | |
| 1:1:1 | 1.0M | 25°C | 2.941 | Present | |
| 1:1:2 | 1.0M | 100°C | 2.900 | Present | |
| 1:1:2 | 1.0M | 50°C | 2.909 | Present | |
| 1:1:2 | 1.0M | 25°C | 2.910 | Present | |
| 3 | 1:1:1 | 1.0M | 100°C | 2.914 | Not present |
| 1:1:1 | 1.0M | 50°C | 2.947 | Not present | |
| 1:1:1 | 1.0M | 25°C | 2.952 | Not present | |
| 1:1:2 | 1.0M | 100°C | 2.886 | Not present | |
| 1:1:2 | 1.0M | 50°C | 2.901 | Not present | |
| 1:1:2 | 1.0M | 25°C | 2.915 | Not present | |
| 4 | 1:1:2 | 1.0M | 100°C | 2.886 | Not present |
| 1:1:2 | 0.50M | 100°C | 2.905 | Not present | |
| 1:1:2 | 0.25M | 100°C | 2.910 | Not present | |
| 1:1:2 | 0.10M | 100°C | 2.933 | Not present | |
| 5 | 1:1:2 | 1.0M | 50°C | 2.901 | Not present |
| 1:1:2 | 0.50M | 50°C | 2.912 | Not present | |
| 1:1:2 | 0.25M | 50°C | 2.919 | Not present | |
| 1:1:2 | 0.10M | 50°C | - | Not present | |
| 6 | 1:1:2 | 1.0M | 100°C | 2.886 | Not present |
| 1:1:2 | 1.0M | 50°C | 2.901 | Not present | |
| 1:1:2 | 1.0M | 25°C | 2.914 | Not present | |
| 7 | 1:1:2 | 0.50M | 100°C | 2.905 | Not present |
| 1:1:2 | 0.50M | 50°C | 2.912 | do- | |
| 1:1:2 | 0.50M | 25°C | 2.919 | Not present | |
| 8 | 1:1:2 | 1.0M | 100°C | 2.900 | Present |
| 1:1:2 | 1.0M | 50°C | 2.909 | Present | |
| 1:1:2 | 1.0M | 25°C | 2.910 | Present | |
| 1:1:2 | 1.0M | 100°C | 2.886 | Not present | |
| 1:1:2 | 1.0M | 50°C | 2.901 | Not present | |
| 1:1:2 | 1.0M | 25°C | 2.915 | Not present | |
| 9 | 1:1:2 | 0.50M | 100°C | 2.902 | Present |
| 1:1:2 | 0.50M | 50°C | 2.910 | Present | |
| 1:1:2 | 0.50M | 25°C | 2.919 | Present | |
| 1:1:2 | 0.50M | 100°C | 2.905 | Not present | |
| 1:1:2 | 0.50M | 50°C | 2.912 | Not present | |
| 1:1:2 | 0.50M | 25°C | 2.919 | Not present |
Figure 4—Effect of increasing concentration. Precipitates were formed from solutions mixed in a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3= at 100°C. Note shift and increase in intensity of major dolomite reflection.
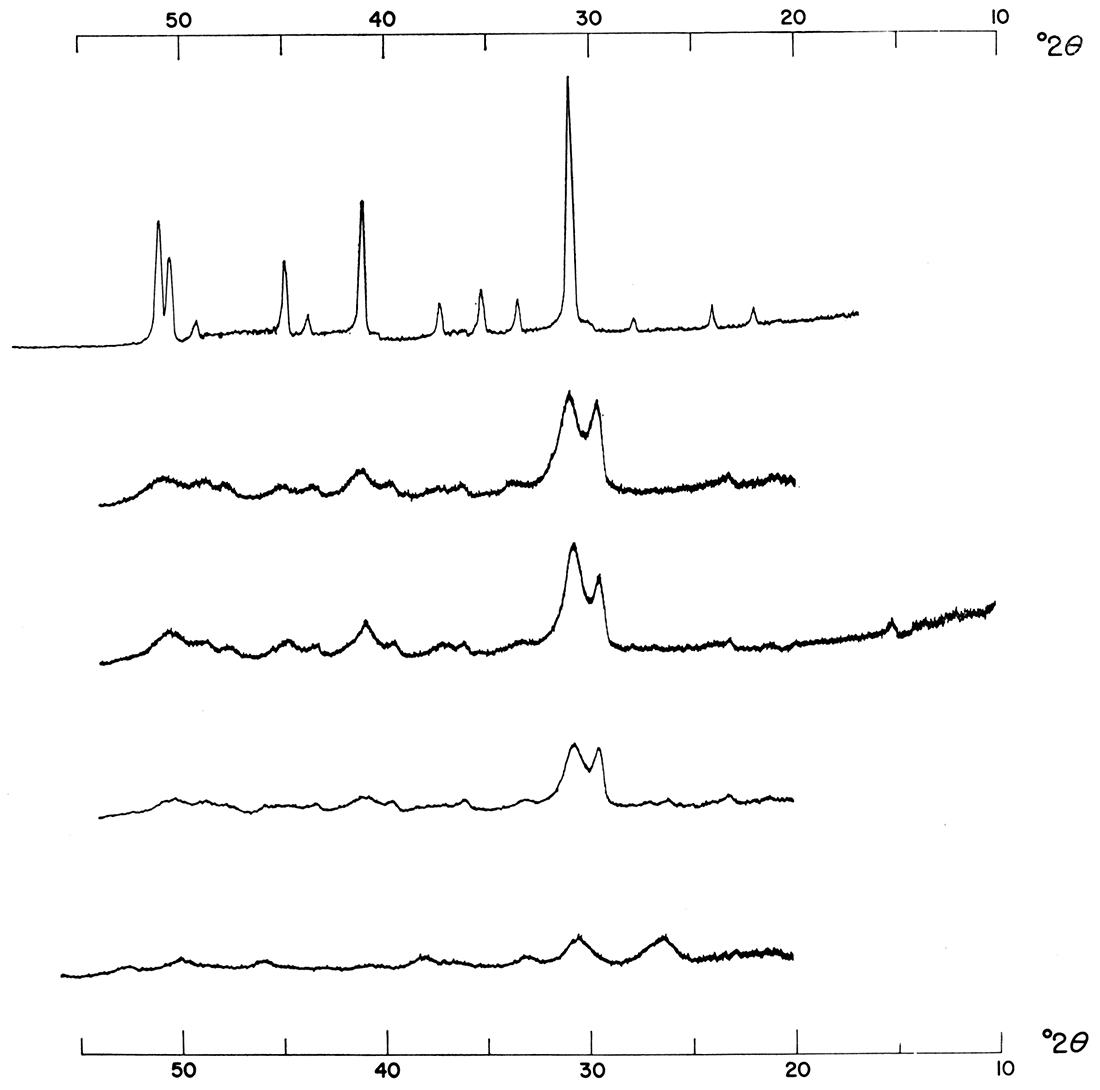
Figure 5—Effect of increasing concentration. Precipitates were formed from solutions mixed in 1:1:2 molar concentration ratio of Ca++: Mg++: CO3= at 50°C. Note shift and increase in intensity of major dolomite reflection.
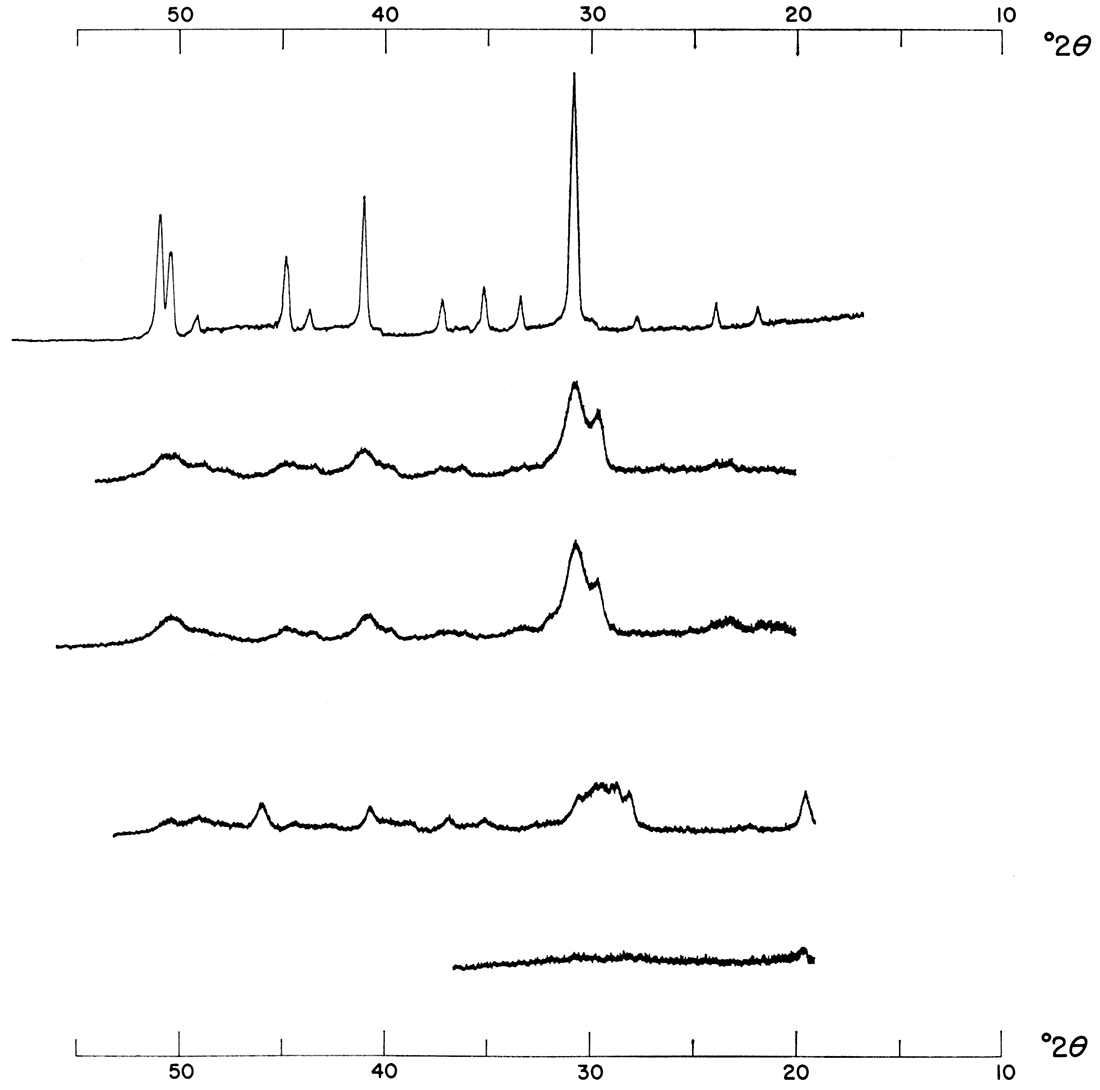
The effects of increasing the temperature of reaction while other conditions remain constant are demonstrated by Figures 6 and 7. Proto-dolomite was precipitated at 25°C, together with calcium sulfate and calcium carbonate, but as the temperature of reaction rose, calcium carbonate was eliminated and there was a shift and increase in intensity of the major dolomite reflection. A higher temperature aids the precipitation of dolomite and eliminates calcium carbonate as a coprecipitate.
Figure 6—Effect of increasing temperature. Precipitates were formed from 1.0 M solutions mixed in a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3=. Note shift and increase in intensity of major dolomite reflection.
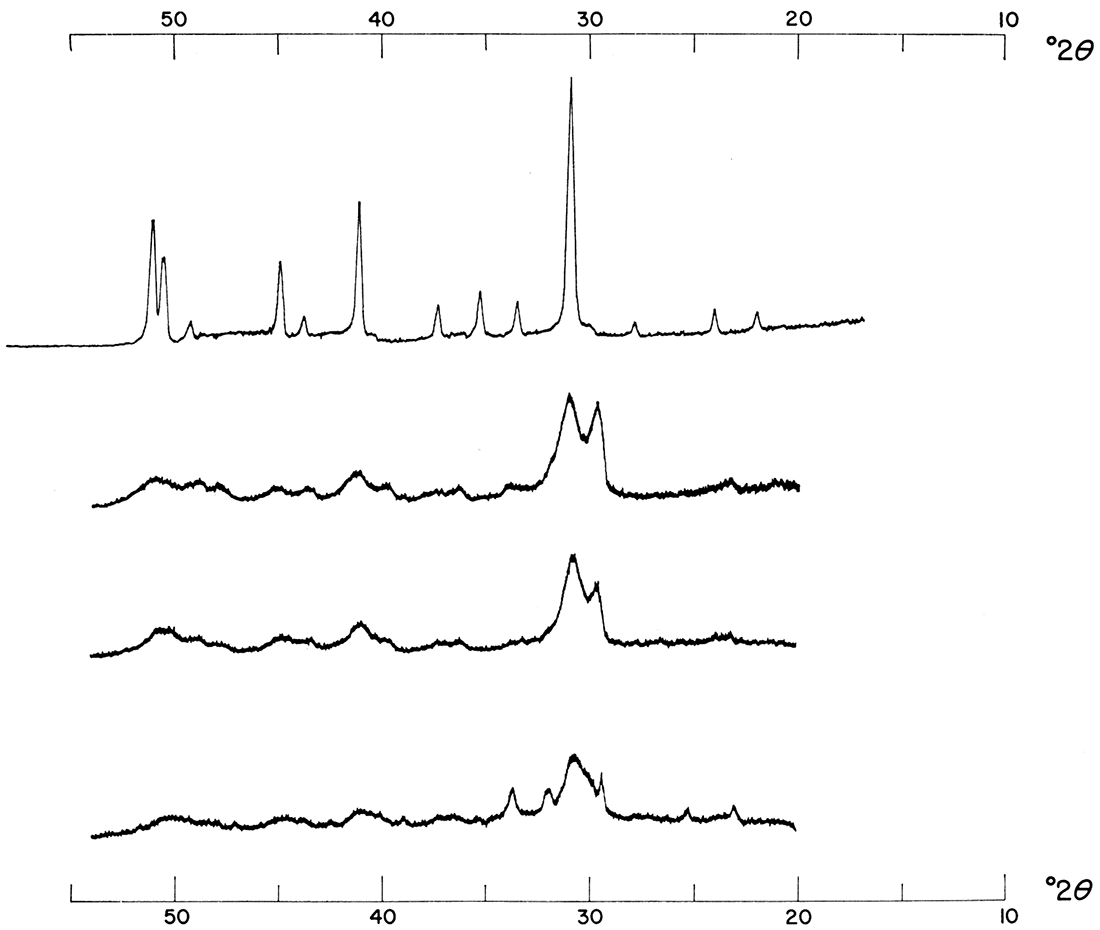
Figure 7—Effect of increasing temperature. Precipitates were formed from 0.5 M solutions mixed in a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3=. Note shift and increase in intensity of major dolomite reflection.
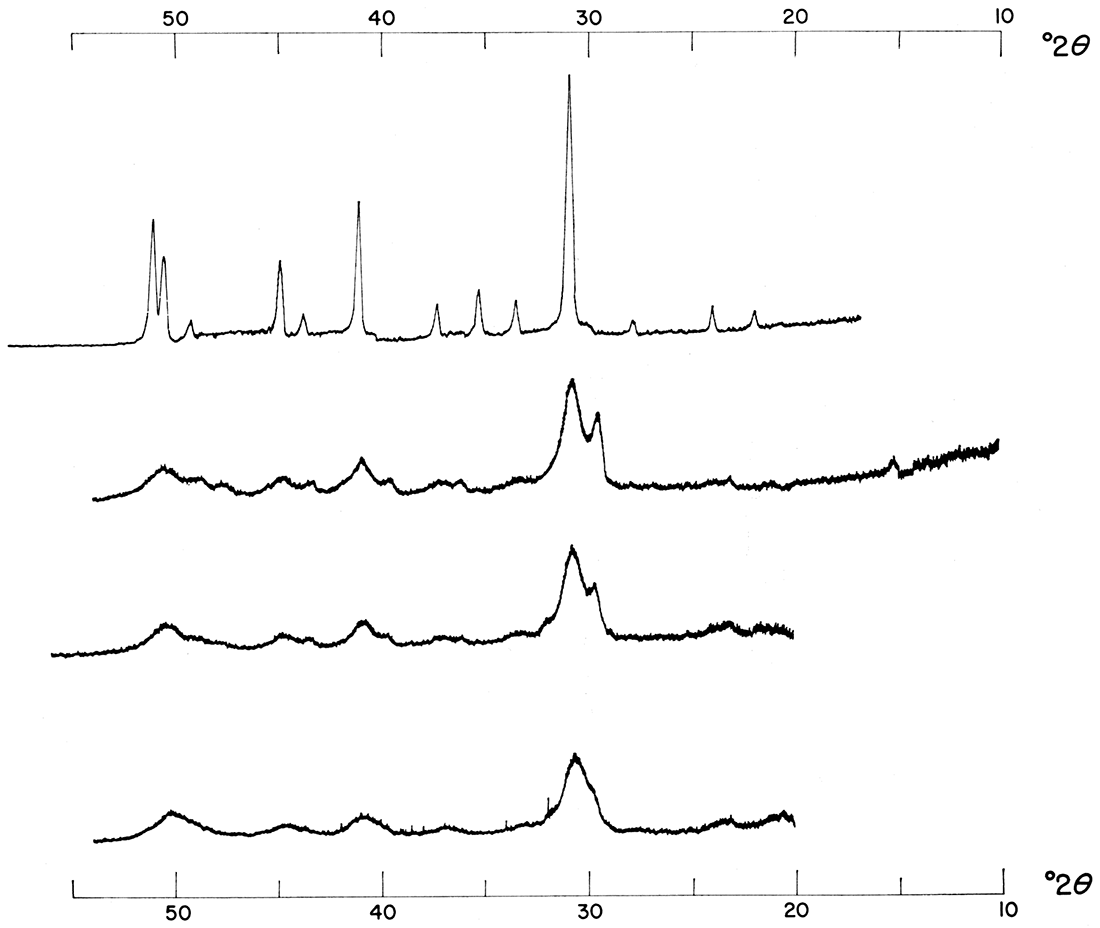
Figure 8 shows the effect of reducing the rate of reaction of precipitations made at various temperatures from 1.0 M solutions mixed in a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3=. At this concentration the presence of activated charcoal in the reacting solutions brings about a product exhibiting better crystallinity. This effect is obvious when the intensities of corresponding products are compared. Precipitations made under the same conditions except that 0.50 M solutions were used did not show the same effect; in fact, the presence of activated charcoal seemed to bring about a slight reduction in crystallinity (Fig. 9). This interpretation is made from a consideration of relative intensities of major dolomite reflections. There were no major peak shifts; therefore a slower rate of reaction did not aid in the ordering of the dolomitic product. When the left portions of Figure 2 and 3 are compared, it is apparent that activated charcoal alters precipitates formed from 1:1:1 molar concentration ratios of Ca++: Mg++: CO3= in the same way as it did the precipitates just described. It is also evident from Figures 2 and 3 that a reduction in the rate of reaction aided the formation of more dolomitic carbonate at the expense of calcium carbonate. It is possible that there is an optimum rate at which perfectly crystalline dolomite forms and at which calcium. carbonate is eliminated in favor of dolomite.
Figure 8—Effect of reducing rate of reaction. Precipitates were formed from 1.0 M solutions mixed in a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3= at various temperatures. Precipitates in top figure were made in the presence of activated charcoal; note increase in intensity of major dolomite reflections of these precipitates, formed at slower rate of reaction.
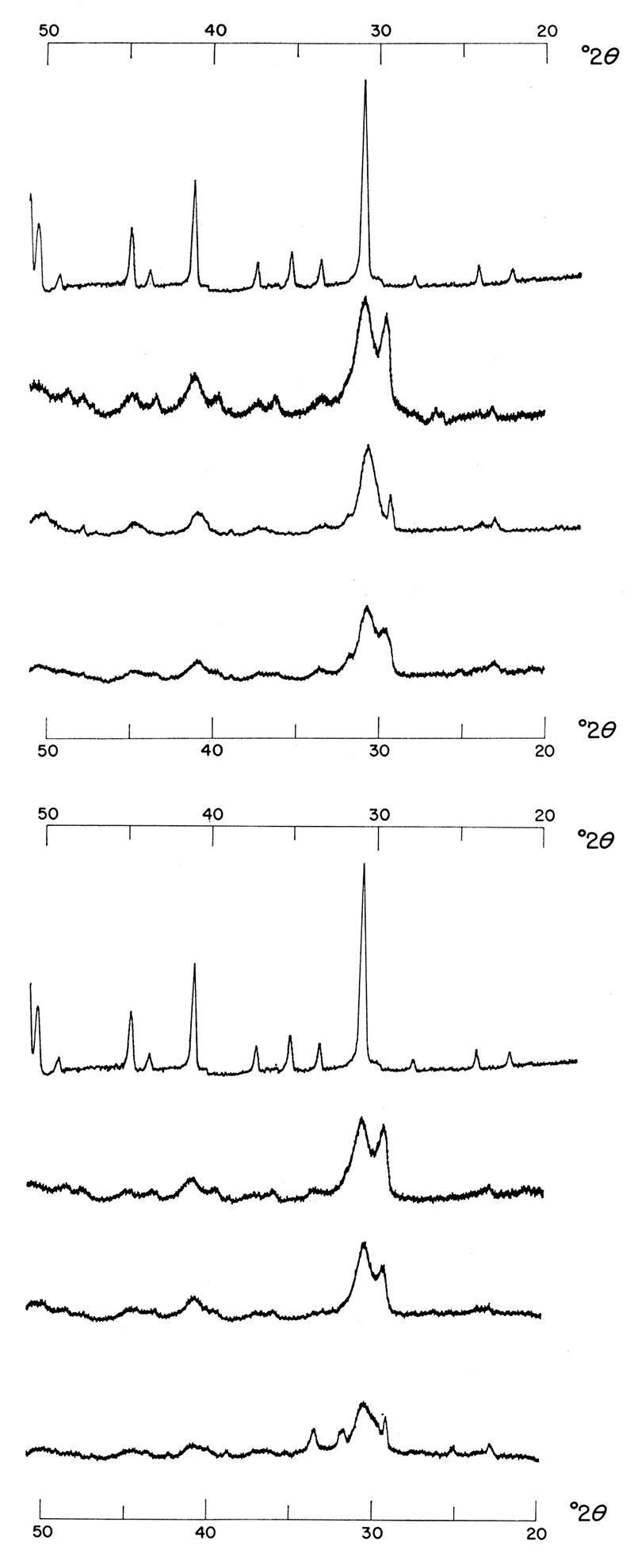
Figure 9—Effect of reducing rate of reaction. Precipitates were formed from 0.5 M solutions mixed in a 1:1:2 molar concentration ratio of Ca++: Mg++: CO3= at various temperatures. Precipitates in top figure were made in the presence of activated charcoal, but at this concentration, change in rate produced no noticeable shift nor intensification of major dolomite reflection.
The origin of primary dolomite has been an enigma to geologists, and the processes that operate in the natural precipitation of that double salt have never been completely understood. Observations made in this study may provide an acceptable explanation for a mechanism of formation of dolomite and for its common association with evaporite environments.
Of the parameters studied in the precipitation of dolomite, the most important are (1) the carbonate ion concentration as it controls the pH of the precipitating medium and (2) the rate of reaction as controlled by the presence of a gel or suspension. An increase in carbonate ion concentration, and the corresponding rise in pH, allows magnesium and calcium ions to coprecipitate to form the double salt in a proto-dolomitic or disordered form depending on temperature or concentration of the reacting solutions. The proposed reactions (Table 3) apply to the formation of dolomite only when a gel or suspension is present during the intermediate reaction. The gel or suspension serves to retard the reaction so that a better crystallinity is observed in the products, and under some conditions it abets the formation of dolomite at the expense of calcium carbonate. It is especially notable that no dolomitic carbonate was produced when finely crystalline reagents of the intermediate products reacted with a solution of sodium carbonate at 100°C; this indicates that the gel or suspension phase might be necessary for formation of dolomite by the technique used in this investigation.
Perhaps the role played by the sulfate ion in the precipitation of dolomite is related to the gel or suspension phase. This role is not evident from experimental data, but the sulfate ion could affect the precipitation process in either of two ways neither of which would occur if the chloride, nitrate, or acetate salts of magnesium were used in lieu of the sulfate salt. First, considering reaction (1) of Table 3, it is observed that a gelatinous calcium sulfate is formed together with n hydrated magnesium gel, which probably can be represented as Mg(H2O)++; these gels could then interact in the presence of high carbonate ion concentration and consequent high pH to form the double salt, CaMg(CO3)2. Second, when the magnesium is added in the form of sulfate, the attendant reaction in which the two gels are formed can cause the cation concentration in the precipitating medium to be' significantly reduced; this would not be the case if the other previously mentioned salts of magnesium were used. Thus the precipitation. takes place from relatively dilute solutions of calcium and magnesium ions in the presence of a high concentration of carbonate ions.
It is certain that the formation of a gel or fine suspension affects the "available" concentration of metal ions and consequently the kinetics of the precipitation. The rate of crystal growth is reduced, and according to theoretical considerations by Kramer (1959) and laboratory observations of this investigation, this reduction in the rate of crystal formation favors the dolomite type structure. C. A. Reynolds, Department of Chemistry, states (personal communication) that a fine suspension of particles, organic or inorganic or combinations thereof, such as might be found in many natural sedimentary environments, would function in the same way as the gel or activated charcoal to retard the reaction.
A situation of this type actually exists in Australia, where Alderman and Skinner (1957) and Alderman (1959) reported that during the spring and summer seasons a fine suspension of sediment is precipitated from the waters of a lagoonal environment and finally settles out as a disordered dolomite. These authors do not use the protodolomite terminology of Graf and Goldsmith (1956), although their product does not show the superlattice reflections necessary for true dolomite by the definition set up in 1956.
It should be emphasized that in this one area of the world where dolomitic carbonate is precipitating directly from sea waters the product is a disordered dolomite or the so-called proto-dolomite. It seems possible that with the passage of time and the attainment of equilibrium, however, true dolomite might be the final mineral phase observed, no trace of proto-dolomite being left. Physical chemists (Daniels and Alberty, 1955) have established the fact that attainment of equilibrium in solid phases is extremely slow, particularly at the low temperatures that exist in near-surface geologic environments. Thus a change in concentration at a surface of a grain of dolomite, for example, would require a geologically significant period of time to affect points in the interior of the grain.
If the uniformitarian concept can be applied to the above observations, the premise can be made, with due reserve, that many of the dolomites in the geologic rock column were originally formed as disordered dolomite but with passage of geologic time they have reached equilibrium and appear as true dolomites. This premise is most likely to be valid for evaporite environments where dolomite occurs. As evaporation progressed and solubility products were exceeded (assuming the presence of necessary elements), a fine suspension of material, perhaps in the form of a gelatinous calcium carbonate, would begin to settle out, increasing the relative concentration of other elements or compounds. If enough magnesium and carbonate ions were available, a reaction similar to that proposed in this study, but probably much more complex, could precipitate a proto-dolomite, with or without the single carbonate phase being present, followed by an ordering, dolomite being the end-product.
Alderman, A. R. (1959) Aspects of carbonate sedimentation: Australia Geol. Soc. Jour., v. 6, pt. 1, p. 1-10.
Alderman, A. R., and Skinner, H. C. W. (1957) Dolomite sedimentation in the South-East of Australia: Am. Jour. Sci., v. 225, p. 561-567.
Baron, Guy (1960) Sur la synthese de la dolomie. Application au phenomene de dolomitization: Inst. Français petrole Rev. et Annales combustibles liquides, no. 1, p. 3-68.
Baron, Guy, and Favre, J. (1959) Recherches experimentales sur le role des facteurs physico-chemique dans la synthese de la dolomie: Paper presented at Fifth World Petroleum Congress.
Cayeux, L. (1935) Les roches sedimentaires de France; Roches carbonatees (calcaires et dolomies): Masson et Cie., Paris, p. 1-463.
Chilingar, G. V. (1956) Relationship between Ca/Mg ratio and geologic age: Am. Assoc. Petroleum Geologists Bull., v. 40, p. 2256-2266.
Clarke, F. W. (1924) The data of geochemistry: U. S. Geol. Survey, Bull. 770, p. 1-1841. [available online]
Cloud, P. E., Jr., and Barnes, V. E. (1946) The Ellenburger Group of Central Texas: Texas Univ. Bull. 4621, p. 89-95.
Daniels, F. (1961) Kinetics and thermoluminescence in geochemistry: Geochem. et Cosmochim. Acta, v. 22, p. 65-74.
Daniels, F., and Alberty, R. A. (1955) Physical chemistry: John Wiley and Sons, Inc., New York, p. 277-317.
De Marignac, C. (1848) Cited in Clarke, 1924, p. 566.
Deville, C. S. (1858) Cited in Clarke, 1924, p. 566.
Durocher, J. (1851) Cited in Clarke, 1924, p. 566.
Fairbridge, R. W. (1957) The dolomite problem, in Regional aspects of carbonate deposition: Soc. Econ. Paleontologists and Mineralogists, Spec. Pub. 5, p. 125-178.
Garrels, R. M. (1960) Mineral equilibria: Harper & Brothers, New York, p. 1-254.
Garrels, R. M., Thompson, M. E., and Siever, R. (1960) Stability of some carbonates at 25°C and 1 atmosphere total pressure: Am. Jour. Sci., v. 258, p. 402-418.
Graf, D. L. (1960) Geochemistry of carbonate sediments and sedimentary carbonate rocks: Illinois Geol. Survey Circ. 297, 298, 301, 308, and 309.
Graf, D. L. and Goldsmith, J. R. (1956) Some hydrothermal syntheses of dolomite and proto-dolomite: Jour. Geology, v. 64, p. 173-186.
Halla, F. (1935) Eine Methode zur bestimmung der Anderung freien Energie bei Reactionen des Typus A(s) + B(s) = AB(s) und ihre Anwendung auf das Dolomitproblem: Zeitschr. Phys. Chemie, Abt. A, v. 175, p. 63-82.
Halla, F. (1936) Bemerkungen zur kongruenten Loslichkeit des Dolomits: Nachtrag zu einer vorangegangenen Arbeit: Phys. Chemie Zeitschr., Abt. A., v. 175, p.396-399.
Hood, D. W., and others (1959) CaCO3 solubility equilibrium in sea water: NSF 1342 and 5038, Ref. 59-13F; Texas A. & M. Projects 109 and 171, College Station.
Hoppe-Seyler, E. (1875) Ueber die Bildung von Dolomit: Deutsch. Geol. Gesell. Zeitschr., v. 27, p. 495-530.
Hunt, T. Sterry (1859) On some reactions of the salts of lime and magnesia and on the formation of gypsums and magnesian rocks: Am. Jour. Sci., ser. 2, v. 28, p. 170-187, 365-383.
Hunt, T. Sterry (1866) Further contributions to the history of lime and magnesia salts: Am. Jour. Sci., ser. 2, v. 42, p. 49-67.
Ingerson, E. (1961, in press) Problems of the geochemistry of sedimentary carbonate rocks: Geochim. et Cosmochim. Acta.
Kramer, J. R. (1959) Correction of some earlier data on calcite and dolomite in sea water: Jour. Sed. Petrology, v. 28, p. 465-467.
Lalou, C. (1957) Studies of bacterial precipitation of carbonates in sea water: Jour. Sed. Petrology, v. 27, p. 190-195.
Lange, N. A., and Forker, G. M. (eds.) (1956) Handbook of chemistry: Handbook Publishers, Inc., Sandusky, p. 1-1969.
Linck, G. (1909) Ueber die Enstehung der Dolomit: Deutsch. Geolog. Gesell. Monatsber., v. 61, p. 230-241.
Linck, G. (1937) Bildung des Dolomits und Dolomitisierung: Chemie der Erde, v. 11, p. 278-286.
Medlin, W. L. (1959) Preparation of synthetic dolomite: Amer. Mineralogist, v. 44, p. 979-986.
Merwin, H. E. (1923) Chemical and physical researches on sediments for the year 1922-1923; Nat. Res. Council, Rept. Comm. Sedimentation, p. 32-37.
Neher, J., and Rohrer, E. (1958) Dolomitbildung unter Mitwirkung von Bacterien: Ecolgae Geologicae Helvetiae, v. 51, no. 2, p. 213-215.
Newell, N. D., and others (1953) The Permian reef complex of the Guadalupe Mountains region, Texas and New Mexico: W. H. Freeman & Company, San Francisco, p. 1-236.
Pauling, L. (1956) General chemistry: W. H. Freeman & Company, San Francisco, 2d ed. (rev.) p. 1-710.
Riviere, A. (1939a) Sur la dolomitisation des sediments calcaires: Paris Acad. Sci., Comptes rendus, v. 209, p. 597-599.
Riviere, A. (1939b) Observations nouvelles sur le mecanisme de dolomitisation des sediments calcaires: Paris Acad. Sci., Comptes rendus, v. 209, p. 691-692.
Riviere, A. (1941) Sur la reserve alcaline et les carbonates de l'eau de mer: Soc. Geol. France, compte rendu sommaire, 5th ser., v. 11, p. 19-20.
Spangenberg, K. (1913) Die kunstliche Darstellung des Dolomits: Zeitschr. f. Kryst. u. Min., v. 52, p. 529-567.
Steidmann, E. (1917) Origin of dolomite as disclosed by stains and other methods: Geol. Soc. America Bull., v. 28, p. 431-450.
Sverdrup, H. U., Johnson, M. W., and Fleming, R. H. (1942) The oceans; their physics, chemistry, and general biology: Prentice-Hall, New York, p. 1-1087.
Twenhofel, W. H., and others (1932) Treatise on sedimentation: 2d ed., Nat. Res. Council, publ. Williams and Williams Company, Baltimore, p. 1-926.
Van Tuyl, F. M. (1916) The origin of dolomite: Iowa Geol. Survey Ann. Rept. 25, p. 251-422.
Von Morlot, A. (1847) Ueber die Dolomit und seine kunstliche Darstellung aus Kalkstein: Haidinger, Nat. Abh., v. 1, p. 305.
Weyl, P. K. (1961) The carbonate saturometer: Jour. Geology, v. 69, p. 32-44.
Zeller, E. J., Saunders, D. F., and Siegel, F. R. (1959) Laboratory precipitation of dolomitic carbonate (abs.): Geol. Soc. America Bull., v. 70, p. 1074.
For the dissociation of dolomite,
CaMg(CO3)2 = Ca++ + Mg++ + 2CO3= (1)
the equation for the solubility product K is:
K= (aCa++) (aMg++) (a2CO3=) (2)
where a is the activity and
aCa++ = 10-2.88; aMg++ = 10-2.87; aCO3= = 10-6.79.
Substituting these values in equation (2),
K= (10-2.88) (10-2.87) (10-6.79)2 =10-19.33.
From the basic thermodynamic relation ΔFR° = -RT ln K = -1.364 log K at 25°C and 1 atmosphere pressure, the standard free energy of reaction for CaMg(CO3)2 = Ca++ + Mg++ + 2 CO3=, is ΔFR° = -1.364 × -19.33 = 26.37 kcal. We may then write:
ΔFR° = ΔFf°Ca++ + ΔFf°Mg++ + 2ΔFf°CO3= - ΔFf°CaMg(CO3)2
Substituting the value for ΔFR° obtained from equation (2), and values for the ions from U.S. Bureau of Mines Circular 500:
+26.37 = -132.18 - 108.99 - 252.44 - ΔFf°CaMg(CO3)2, and transposing, ΔFf°CaMg(CO3)2 = -519.99 kcal.
A check on this value for the free energy of formation of dolomite can be made using the relationship that exists when dolomite dissolves to equilibrium in water:
CaMg(CO3)2 + 2H2O + 2CO2 = Ca++ + Mg++ + 4 HCO3-; K = 10-14.31
ΔFR° = +19.52 kcal.
We may now write:
+19.52 = ΔFf°Ca++ + ΔFf°Mg++ + 4ΔFf°HCO3- - 2ΔFf°CaMg(CO3)2 - 2ΔFf°H2O - 22ΔFf°CO2
Substituting as above: +19.52 = -132.18 -108.99 - 561.24 - ΔFf°CaMg(CO3)2 + 113.4 + 188.52, and,
ΔFR°CaMg(CO3)2 = -520.00 kcal.
(1) Ca++ + 2NO3- + Mg++ + SO4= → α-CaSO4; H2O + Mg++ + 2NO3-
ΔFR° = ΔFf°α-CaSO4 · 1/2 H2O + ΔFf°Mg++ + 2ΔFf°NO3- - ΔFf°Ca++ - 2ΔFf°NO3- - ΔFf°Mg++ - ΔFf°SO4=
ΔFR° = -343.02 - 108.99 - 2(26.43) + 132.18 +2 (26.43) + 108.99 + 177.34
ΔFR° = -33.50 kcal
(2) α-CaSO4 · 1/2 H2O + Mg++ + 2NO3- + 4Na+ + 2CO3= → CaMg(CO3)2 + α-CaSO4 · H2O + 4Na+ + 2NO3-
ΔFR° = ΔFf°CaMg(CO3)2 + ΔFf°α-CaSO4 · 1/2 H2O + 4ΔFf°Na+ + 2ΔFf°NO3- - ΔFf°α-CaSO4 · 1/2 H2O - ΔFf°Mg++ - ΔFf°NO3- - ΔFf°Na+ - 2ΔFf°CO3=
ΔFR° = -520.0 - 343.02 - 4 (62.59) -2 (26.43) + 343.02 + 108.99 + 2 (26.34) + 4 (62.59) + 2 (126.22)
ΔFR° = -158.57 kcal.
ΔFcoupled reactions = -192.07 kcal
(1) Ca++ + 2NO3- + Mg++ + SO4= → CaSO4 + Mg++ + 2NO3-
ΔFR° = ΔFf°CaSO4 + ΔFf°Mg++ + 2ΔFf°Mg++ - ΔFf°Ca++ - 2ΔFf°NO3- - ΔFf°Mg++ -ΔFf°SO4=
ΔFR° = -3l2.46 - 108.99 - 2 (26.43) + 132.18 + 2 (26.43) + 108.99 + 177.34
ΔFR° = -2.94 kcal
(2) CaSO4 + Mg++ + 2NO3- + 4Na+ + 2CO3= → CaMg(CO3)2 + CaSO4 + 4Na+ + 2NO3-
ΔFR°= ΔFf°CaMg(CO3)2 + ΔFf°CaSO4 +4ΔFf°Na+ + 2ΔFf°NO3- - ΔFf°CaSO4 - ΔFf°Mg++ - 2ΔFf°NO3- - 4ΔFf°Na+ - 2ΔFf°2CO3=
ΔFR° = -520.0 - 312.46 - 4(62.59) - 2(26.43) + 312.46 + 108.99 + 2(26.43) + 4(62.59) + 2(126.22)
ΔFR° =-158.57 kcal
ΔFcoupled reactions = -161.51 kcal
The following chemical relations are valid:
From Kramer, 1959
From Garrels, 1960, p. 45
For the calculations that follow, four premises must be made:
[H2CO3] / pCO2 = KCO2 = 10-1.5
[H2CO3] = 10-1.5 × pCO2
If pCO2 = 10-3.5 (normal atmospheric pressure),
[H2CO3] = 10-1.5 × 10-3.5 = 10-5
From chemical relation 2,
[H+] [HCO3-] = 10-6.4 × 10-5 = 10-11.4; transposing
[HCO3-] = 10-11.4 / [H+]
From chemical relation 3,
[CO3=] = [10-10.3] [HCO3-] / [H+]
substituting for [HCO3-],
[CO3"] = 10-21.7 [H+]2
From chemical relation 4,
[OH-]= 10-14 [H+]
From chemical relation 1,
[Ca++] [Mg++] = 10-19-33 / [CO3=]2
[Ca++] [Mg++] = 10-19-33 / {10-21.7 / [H+]2}2 = 1024.07 [H+]4
On dissolution of dolomite,
CaMg(CO3), ↔ Ca++ + Mg++ + 2(CO3)=,
we can assume that the ion activities are essentially equal to their molalities and that the dolomite will dissolve congruently so that [Ca++] ≐ [Mg++],
Hence, we can write:
[Ca++] ≐ [Mg++] ≐ (1024.07[H+]4)1/2 ≐ 1012.035 [H+]2
Now, substituting in the expression for electrical neutrality.
4 × 1012.035 [H+]2 + [H+] = 2 × (10-21.7 / [H+]2) + (10-11.4 / [H+]) + (10-14 / [H+])
Multiply through by [H+]2 gives:
4 × 1012.035 [H+]4 + [H+]3 = 2 × 10-21.7 + 10-11.4 [H+] + 10-14 [H+]
Regroup:
1012.642 [H+]4 + [H+]3 - 10-11.399 [H+] = 10-21.399
Solve by trial and error, [H+] = 107.57
∴ pH ≐ 7.6
If the same calculation is made. but with pCO2=10-1.3 (158 times normal), the values for [H2CO3], [HCO3-], [Ca2+], [Mg2+], and [CO3=] are recalculated with the pCO2, as:
[H2CO3] = 10-2.3
[CO3=] = 10-19.5 / [H+]
[HCO3-] = 10-9.2 / [H+]
[Ca++] ≐ [Mg++] ≐ 109.835
Substitution into the expression for electroneutralily and multiplying by [H+]3 gives:
4 × 109.835 [H+]4 + [H+]3 = 2 × 10-19.5 + 10-9.2 [H+] + 10-14 [H+]
Regrouping and trial and error solution gives:
[H+] ≐ 10-6.47 and pH ≐ 6.5
These data demonstrate that an increase of 158 times in the partial pressure of carbon dioxide decreases the pH of a distilled water medium in equilibrium with dolomite about 1.1 pH units. If the self-contained buffering system of sea water is considered, it is probable that an increase of the same magnitude for pCO2 as noted previously would not greatly affect the pH of sea water, and carbonate deposition would not be significantly affected.
Kansas Geological Survey
Placed on web Dec. 12, 2018; originally published Dec. 31, 1961.
Comments to webadmin@kgs.ku.edu
The URL for this page is http://www.kgs.ku.edu/Publications/Bulletins/152_5/index.html